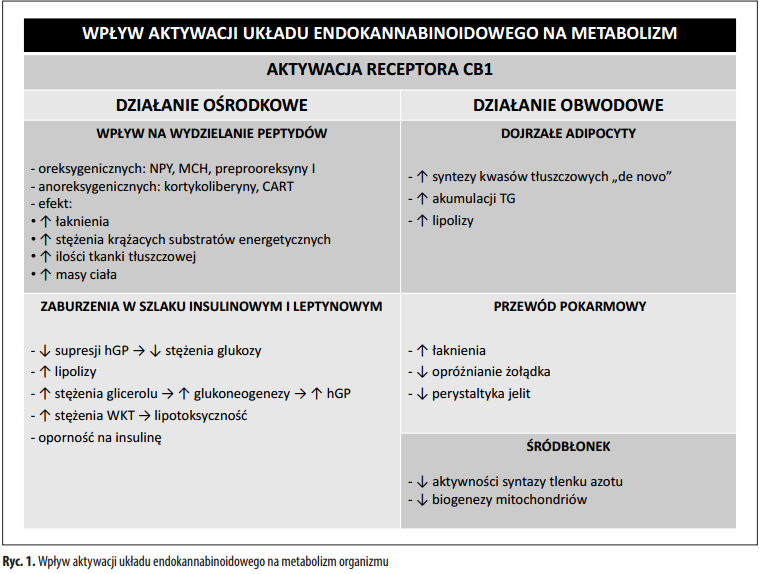
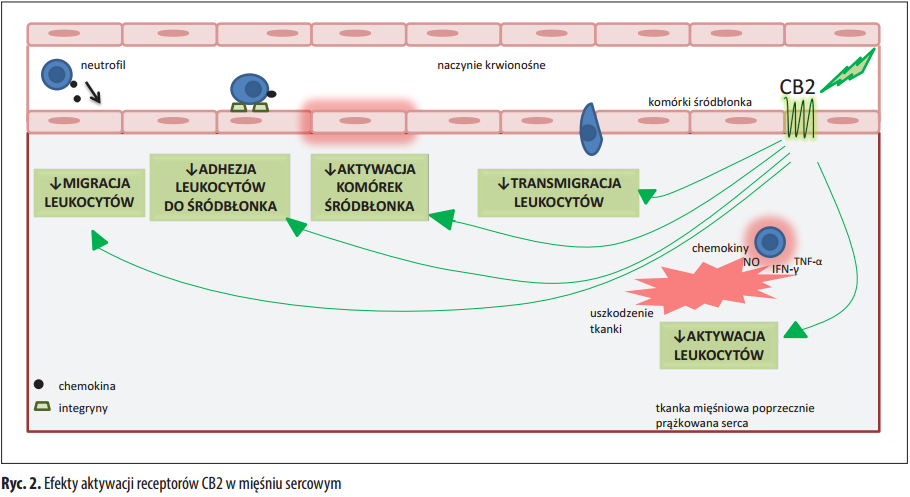
W ciągu ostatnich kilku lat byliśmy świadkami wzrostu liczby nowych doniesień dotyczących roli kannabinoidów, zarówno syntetycznych, jak i ziołowych, w mechanizmach zapalenia i karcynogenezy. Jednakże pomimo bogactwa danych in vitro i niepotwierdzonych doniesień, brakuje dowodów na to, że kannabinoidy mogą działać jako korzystne leki w leczeniu nieswoistego zapalenia jelit (IBD) lub rozwoju nowotworu przewodu pokarmowego u człowieka. Pewien wgląd w wpływ marihuany medycznej (zwykle oznaczającej suszone kwiaty) i kannabinoidów na IBD uzyskano dzięki kwestionariuszom i małym badaniom pilotażowym. Jeśli chodzi o raka jelita grubego, dostępne są jedynie dane przedkliniczne. Obecnie Δ<sup>9</sup>-tetrahydrokannabinol (THC) i jego syntetyczne formy, dronabinol i nabilon, stosuje się jako leczenie uzupełniające w celu łagodzenia przewlekłego bólu i spastyczności u pacjentów ze stwardnieniem rozsianym, a także nudności wywołanych chemioterapią . Używanie marihuany medycznej jest dozwolone tylko w ograniczonej liczbie krajów. Żadna z wymienionych substancji nie jest obecnie wskazana w leczeniu IBD. Niniejsza recenzja stanowi aktualizację naszej wiedzy na temat roli kannabinoidów w zapaleniu jelit i karcynogenezie oraz dyskusję na temat ich potencjalnego zastosowania terapeutycznego.
Korzystne właściwości kannabinoidów w przewodzie żołądkowo-jelitowym (GI) wynikają z faktu, że jelita są wyposażone w układ endokannabinoidowy (ECS), sieć regulacyjną receptorów kannabinoidowych, enzymów i ligandów, które odgrywają kluczową rolę w procesach fizjologicznych, jak i fizjologicznych. procesy patofizjologiczne [ 1 , 2 , 3 , 4 ]. Tak więc w jelitach występuje ekspresja klasycznych receptorów kannabinoidowych 1 i 2 (CB 1 , CB 2 ) oraz niereagujących na kannabinoidy receptorów CB 1 /CB 2 (receptor sprzężony z białkiem G 55 [GPR55], członek podrodziny kanałów kationowych o potencjale przejściowym 1 [TRPV1] oraz receptory gamma i alfa aktywowane przez proliferatory peroksysomów [PPARγ i PPARα]). Ponadto enzymy wytwarzające endokannabinoidy (EC), takie jak specyficzna dla N-acylofosfatydyloetanoloaminy fosfolipaza D i lipaza diacyloglicerolu, oraz enzymy rozkładające EC, takie jak hydrolaza amidów kwasów tłuszczowych (FAAH) i lipaza monoacyloglicerolu (MGL), znajdują się w jelito. CB 1 i CB 2 lokalizują się głównie w nerwach jelitowych, nabłonku jelitowym i komórkach odpornościowych o zmiennej ekspresji [ 5 , 6 , 7 ]. Podczas gdy CB 1 ulega ekspresji na wysokim poziomie w cholinergicznych neuronach jelitowych [ 8 ], CB 2 ulega dużej ekspresji w komórkach odpornościowych [ 9 ].
Anandamid (AEA) i 2-arachidonoiloglicerol (2-AG) to najlepiej opisane EC, które aktywują receptory CB 1 , CB 2 i wyżej wymienione receptory inne niż CB 1 /CB 2 [ 10 , 11 , 12 ]. Oprócz tych składników ECS w przewodzie pokarmowym znaleziono lipidy podobne do EC, głównie N-acyloetanoloaminy, takie jak palmitoiloetanoloamid (PEA) i oleiloetanoloamid. Tam działają na receptory inne niż CB 1 /CB 2 , takie jak GPR55 i GPR119 i wpływają na sygnalizację AEA, zwaną także „efektem otoczenia” [ 13 ]. EC są najprawdopodobniej zaangażowane w liczne mechanizmy regulacyjne, np. utrzymują nienaruszoną barierę nabłonkową [ 14 , 15 ] i utrzymują tolerancję immunologiczną poprzez kontrolowanie ekspansji regulatorowego podzbioru limfocytów T Tr1 i obecność immunosupresyjnych makrofagów CX3CR1hi [ 16 ] . Nieoczekiwanie donoszono o zmianach w poziomach EC i lipidów podobnych do EC u pacjentów z nieswoistym zapaleniem jelit (IBD) [ 17 ] i rakiem jelita grubego (CRC) [ 18 ]. Nie wiadomo jednak, czy zmiany te rzeczywiście korelują z postępem choroby. Jeśli chodzi o CRC, dane in vitro z linii komórkowych raka okrężnicy przekonująco pokazują antyproliferacyjne działanie kannabinoidów [ 19 ] i w rzeczywistości modele CRC u myszy z nokautem sugerują antyonkogenną rolę co najmniej CB 1 [ 20 , 21 ]. Jednakże w odniesieniu do CB 2 badania na ludzkich pacjentach z CRC pokazują, że jego ekspresja koreluje ze zmniejszonym przeżyciem [ 22 ]. W podobny sposób wykazano, że ekspresja CB 1 koreluje z gorszym współczynnikiem przeżycia w stabilnym mikrosatelitarnym CRC w stadium II [ 23 ]. Dlatego doniesienia z badań na ludziach i eksperymentów są kontrowersyjne co do korzystnej roli ECS w CRC. W poniższych rozdziałach pokrótce podsumowano najnowsze wyniki dotyczące roli ECS i działania kannabinoidów w IBD i CRC, po czym nastąpiła dyskusja na temat potencjalnej terapii kannabinoidami.
Kannabinoidy i IBD
IBD, którego głównymi objawami są wrzodziejące zapalenie jelita grubego i choroba Leśniowskiego-Crohna (CD), charakteryzuje się przewlekłymi i nawracającymi atakami zapalnymi przewodu pokarmowego [ 24 ]. Chociaż szczegółowe mechanizmy są nadal nieznane, niekontrolowana i źle ukierunkowana odpowiedź immunologiczna przeciwko antygenom drobnoustrojów w połączeniu z predyspozycjami genetycznymi i czynnikami środowiskowymi przyczynia się do wieloczynnikowego pojawienia się choroby [ 24 , 25 ].
Spostrzeżenia z modeli zwierzęcych
Dane funkcjonalne służące do badania roli receptorów kannabinoidowych i ECS w IBD uzyskano głównie z modeli zwierzęcych i wykazały one, że składniki ECS ulegają zmianie podczas eksperymentalnego zapalenia jelit. Zatem doniesienia o podwyższonych poziomach CB 1 , CB 2 i AEA sugerują zwiększoną sygnalizację kannabinoidową w stanach zapalnych [ 26 , 27 , 28 ]. Farmakologiczna aktywacja CB 1 i CB 2 łagodzi eksperymentalne zapalenie jelita grubego [ 28 , 29 , 30 ], natomiast stosowanie antagonistów lub ablacja genetyczna receptorów kannabinoidowych nasila stan zapalny [ 26 , 31 ]. Co więcej, genetyczny niedobór FAAH lub hamowanie FAAH lub MGL ( a w konsekwencji wzrost AEA lub 2-AG) okazały się chronić przed zapaleniem okrężnicy wywołanym przez siarczan dekstranu sodu (DSS) lub kwas trinitrobenzenosulfonowy (TNBS) [ 31,32 , 33 , 34 , 35 , 36 ].
Kilka fitokannabinoidów, a także syntetycznych analogów i związków podobnych do EC wykazało korzystne działanie w zwierzęcych modelach zapalenia jelit. Psychotropowe Δ 9 -tetrahydrokannabinol (THC) i niepsychotropowy kannabidiol (CBD) zmniejszały zapalenie jelita grubego wywołane TNBS w okrężnicy szczura [ 37 ]. W przeciwieństwie do THC, CBD ma wyjątkowo niskie powinowactwo do CB 1 i CB 2 , ale wykazano, że ma antagonistyczne działanie na GPR55 [ 11 ]; Opisano, że CBD działa na PPARγ [ 38 ] i TRPV1 oraz hamuje aktywność FAAH, zmieniając w ten sposób poziomy EC [ 39 , 40 ]. Wykazano także, że wywiera działanie ochronne w modelu zapalenia okrężnicy wywołanego kwasem dinitrobenzenosulfonowym (DNBS) u myszy [ 41 ]. Nowsze prace wykazały, że ekstrakt z Cannabis sativa o wysokiej zawartości CBD, a nie samo CBD, zapewnia ochronę [ 34 ]. Wykazano , że analog CBD O-1602, który również nie ma powinowactwa do CB 1 i CB 2 , ale ma właściwości agonistyczne w stosunku do GPR55 [ 11 ], ma działanie przeciwzapalne w zapaleniu jelita grubego; jednakże nie pośredniczył w nich GPR55 [ 42 ]. Wydaje się, że O-1602 pośredniczy w zmniejszaniu motoryki okrężnicy poprzez GPR55 [ 43 ]. Prace w naszym własnym laboratorium wykazały, że GPR55 może mieć działanie prozapalne, ponieważ antagonista GPR55 i genetyczna delecja genu GPR55 złagodziły zapalenie jelita grubego DSS u myszy [ 44 ]. Wykazano, że kannabigerol, niepsychotropowy fitokannabinoid, łagodzi zapalenie okrężnicy u myszy i zmniejsza wytwarzanie tlenku azotu w makrofagach oraz powstawanie reaktywnych form tlenu w komórkach nabłonka jelitowego [ 45 ]. Innym przeciwzapalnym i niepsychotropowym fitokannabinoidem jest kannabichromen, który, jak wykazano, hamuje inaktywację EC [ 46 ].
Syntetyczny, nieselektywny agonista CB 1 /CB 2 WIN 55,212-2 został niedawno przetestowany w leczeniu zapalenia jelita grubego wywołanego DSS i wykazał właściwości ochronne i przeciwzapalne, które wydają się być przynajmniej częściowo pośredniczone przez hamowanie kinazy białkowej aktywowanej mitogenem p38 [ 47 ] . Innym nieselektywnym agonistą receptora kannabinoidowego ostatnio testowanym w leczeniu zapalenia jelita grubego wywołanego DSS jest HU210 [ 48 ]. Wcześniej wykazano, że ma działanie ochronne w modelu zapalenia okrężnicy wywołanego DNBS [ 26 ], substancja ta utrzymuje integralność funkcji bariery jelitowej niezależnie od receptora Toll-podobnego 4, ale wywołuje pozajelitowe działanie przeciwzapalne zależne od receptora Toll-podobnego 4- za pośrednictwem aktywacji kinazy białkowej p38 aktywowanej mitogenem [ 48 ].
Związek podobny do EC, PEA, jest wytwarzany endogennie w wyniku urazu zapalnego i opisano, że działa poprzez CB1 , CB2 , GPR55, PPARα i TRPV1 [ 49 ]. W kilku modelach chemicznie wywołanego zapalenia okrężnicy u myszy wykazano, że PEA zmniejsza stan zapalny [ 49 , 50 ] i chroni przepuszczalność jelit przez PPARα, CB2 i GPR55 [ 49 ]. Zgodnie z tymi wynikami, hamowanie enzymu degradującego PEA, N-acyloetanoloaminy, hydrolizującej kwaśnej amidazy, zwiększało poziomy PEA, a także chroniło przed zapaleniem jelita grubego [ 51 ]. Ponadto Sarnelli i in. [ 52 ] podali, że hamowanie angiogenezy związanej ze stanem zapalnym przez PEA w modelu DSS było zależne od PPARα. Dodatkowo wykazali, że uwalnianie czynnika wzrostu śródbłonka naczyniowego i tworzenie naczyń uległy zmniejszeniu, co doprowadziło do zmniejszenia uszkodzenia błony śluzowej [ 52 ]. Niedawno odkryto, że analog PEA, adelmidrol, wywiera działanie przeciwzapalne, w którym częściowo pośredniczy PPARγ [ 53 ].
Podsumowując, chociaż nadal brakuje szczegółowych mechanizmów działania wszystkich tych związków, dane przedkliniczne są obiecujące i sugerują, że syntetyczne i ziołowe kannabinoidy, inhibitory FAAH i MGL (a w konsekwencji zwiększone poziomy EC i lipidów EC) mogą być przydatne w leczeniu leczyć IBD. Niemniej jednak znalezienie ukierunkowanego leczenia, szczególnie do długotrwałego stosowania i wywołującego niewielkie skutki uboczne, będzie trudne, biorąc pod uwagę, że ECS jest obecny w różnych tkankach całego organizmu i bierze udział w wielu procesach fizjologicznych.
Kannabinoidy i CRC
Obserwacje zmienionych poziomów ekspresji składników ECS w biopsjach nowotworów wskazują na kluczową rolę ECS w rozwoju CRC. W zmianach CRC zwiększył się poziom AEA i 2-AG oraz enzymów odpowiedzialnych za syntezę i degradację EC, co wskazuje na zwiększony metabolizm EC [ 18 , 54 , 55 ]. Jednakże, w porównaniu z sąsiadującą nienowotworową błoną śluzową okrężnicy, stwierdzono, że ekspresja CB 1 jest obniżona w próbkach CRC w wyniku hipermetylacji DNA wysp CpG w regionie promotora CNR1 (gen kodujący CB 1 ) [ 20 , 21 , 56 ] . W ciągu ostatnich lat opublikowano mnóstwo badań przedklinicznych, w których opisano możliwe właściwości przeciwnowotworowe (endo)kannabinoidów, w tym właściwości antyproliferacyjne, proapoptotyczne, antyangiogenne, przeciwmigracyjne i przeciwinwazyjne. Mechanizmy molekularne, dzięki którym kannabinoidy wywierają działanie przeciwnowotworowe, zostały bardzo szczegółowo podsumowane w innym miejscu [ 57 , 58 , 59 , 60 ]. Chociaż dane z testów komórkowych wydają się bardzo obiecujące, dane uzyskane z modeli in vivo są skąpe i tworzą dość skomplikowany obraz. Na przykład leczenie nieselektywnym agonistą CB1 / CB2 HU210 lub CBD zmniejszyło rozwój zmian przednowotworowych w mysich modelach chemicznie wywołanego CRC [ 61 , 62 ]. Podobne wyniki uzyskano zwiększając poziom EC poprzez hamowanie enzymów degradujących FAAH i MGL [ 61 , 63 ]. Jednakże inne badanie wykazało, że blokada sygnalizacji ECS przez zastosowanie antagonisty CB1 SR141716 również zmniejszyła powstawanie zmian przednowotworowych w mysim modelu CRC [ 64 ].
Ostatnio wyszło na jaw, że GPR55, nietypowy receptor kannabinoidowy reagujący na niektóre (endo)kannabinoidy, wywiera działanie sprzyjające rozwojowi nowotworu poprzez zwiększenie masy guza i przerzutów u myszy [ 21 , 65 ]. Podkreśla to pogląd, że ECS i powiązane z nim składniki pełnią różnorodne i często przeciwstawne funkcje oraz że niezbędne jest lepsze zrozumienie podstawowych mechanizmów i interakcji receptorów. Na przykład konieczne będzie uzyskanie większej ilości informacji na temat ich wpływu na populację komórek odpornościowych związaną z nowotworami, tj. mikrośrodowisko nowotworu, zanim kannabinoidy będą mogły zostać przeniesione do kliniki. Ponieważ immunoterapię nowotworową wprowadza się obecnie nawet w przypadku guzów litych [ 66 ], dość niepokojące jest to, że jak dotąd tylko kilka badań dotyczyło wpływu kannabinoidów na mikrośrodowisko guza. Doniesiono, że WIN 55,212-2 (nieselektywny agonista CB1 / CB2 ) i JWH133 (selektywny agonista CB2 ) mają wyraźniejsze działanie przeciwnowotworowe na ksenoprzeszczepy ludzkiego czerniaka u myszy z prawidłową odpornością niż na te wszczepione myszom SCID [ 67 ], co wskazuje, że działanie przeciwnowotworowe (endo)kannabinoidów odbywa się częściowo za pośrednictwem komórek odpornościowych. Jednak inne badanie wykazało, że THC hamuje reaktywność immunologiczną gospodarza przeciwko rakowi płuc u myszy, promując w ten sposób wzrost nowotworu [ 68 ]. To odkrycie zostało potwierdzone przez Hegde i in. [ 69 ], którzy podali, że podawanie THC spowodowało masową ekspansję komórek supresorowych pochodzących z szpiku, heterogennej populacji komórek o silnych właściwościach immunosupresyjnych. Dodatkowo niedawno wykazaliśmy, że skład komórek odpornościowych obecnych w nowotworach jelita grubego u myszy z niedoborem GPR55 był zmieniony w porównaniu z myszami typu dzikiego [ 21 ].
Podsumowując, wydaje się, że wpływ (endo)kannabinoidów na nowotwory jest znacznie bardziej zróżnicowany, niż obecnie szacuje się na podstawie obiecujących danych in vitro. Dlatego możemy potrzebować bardziej dokładnych badań podstawowych na temat interakcji ECS z przedziałem odpornościowym nowotworu przed rozpoczęciem badań klinicznych z kannabinoidami lub medyczną marihuaną jako metodą leczenia przeciwnowotworowego.
Czy kannabinoidy są opcją terapeutyczną w przypadku zapalenia i nowotworu przewodu pokarmowego?
Konopie indyjskie /kannabinoidy w IBD
Zapalenie jelit jest wysoce podatne na leczenie kannabinoidami, co udokumentowano w przypadku IBD u ludzi nie tylko niepotwierdzonymi doniesieniami, ale, co ważne , także kilkoma badaniami kwestionariuszowymi [ 70,71,72,73 ] , a także badaniami obserwacyjnymi i prospektywnymi badaniami klinicznymi [ 70,71,72,73 ] , 74 , 75 , 76 , 77 ]. Wiele kwestionariuszy ujawniło, że pacjenci z IBD często samoleczą się konopiami indyjskimi, aby złagodzić ból brzucha i biegunkę. Jednak długotrwałe używanie konopi indyjskich nie zawsze może być korzystne. Ankieta przeprowadzona przez Storr i in. [ 72 ] wykazali, że używanie konopi indyjskich przez ponad 6 miesięcy było silnym czynnikiem predykcyjnym konieczności przeprowadzenia operacji u pacjentów z CD. Badania prospektywne również wykazały mieszane wyniki dotyczące skuteczności leczenia kannabinoidami u pacjentów z IBD. Podczas gdy małe badanie z udziałem 21 pacjentów z CD wykazało, że krótkotrwałe stosowanie (8 tygodni) konopi indyjskich bogatych w THC spowodowało obniżenie wskaźników aktywności choroby Leśniowskiego-Crohna u prawie wszystkich pacjentów [ 76 ], inne badanie wykazało, że 2-miesięczne leczenie w umiarkowanej CD z niepsychoaktywnym CBD nie było korzystne [ 78 ]. Dodatkowo przeprowadzono eksperymenty na ludzkich tkankach okrężnicy, które dostarczyły informacji na temat przydatności terapii IBD opartej na kannabinoidach. Wykorzystując próbki tkanek okrężnicy od pacjentów z IBD i wyrostków robaczkowych, Couch i in. [ 79 ] wykazali, że inkubacja z CBD i PEA zapobiegała wytwarzaniu cytokin zapalnych odpowiednio poprzez szlaki CB2 i TRPV1 oraz poprzez PPARα. Inne badanie z użyciem eksplantatów od pacjentów z IBD wykazało, że ekstrakt z konopi indyjskich tłumił ekspresję cyklooksygenazy-2 i metaloproteinazy-9 [ 80 ]. Autorzy opisali kwas Δ 9 -tetrahydrokannabinolowy jako aktywny, niepsychotropowy składnik przeciwzapalny ekstraktu, który może być przydatny w leczeniu IBD zamiast CBD.
Podsumowując, badania te sugerują, że leczenie konopiami indyjskimi i kannabinoidami rzeczywiście może złagodzić stan zapalny w IBD, ale najprawdopodobniej zależy to od dawki, sposobu stosowania, a w konsekwencji stężenia w tkance, które można osiągnąć za pomocą kannabinoidów. Jednak wadą jest to, że leczenie kannabinoidami aktywującymi receptor CB 1 wiąże się z psychotropowymi skutkami ubocznymi. THC i jego pochodne mogą powodować zawroty głowy, senność, euforię i halucynacje w wyniku aktywacji CB 1 w mózgu [ 81 ]. Problem ten można obejść, stosując kannabinoidy działające obwodowo, co wydaje się wysoce prawdopodobne, biorąc pod uwagę znaczną obecność ECS w jelitach. Jednakże na motorykę przewodu pokarmowego duży wpływ ma ośrodkowa CB 1 [ 82 ], a obwodowo ograniczeni agoniści CB 1 /CB 2 podani dootrzewnowo nie poprawili stanu zapalnego w eksperymentalnych modelach zapalenia jelit [ 83 , 84 ]. Zamiast tego działały ochronnie, gdy były stosowane do komory mózgowej [ 84 ]. Ponadto niedobór CB 1 w nerwie błędnym spowalnia motorykę przewodu pokarmowego [ 85 ]. Obserwacje te wskazują, że obwodowa aktywacja receptorów kannabinoidowych może nie być wystarczająca do złagodzenia stanu zapalnego przewodu pokarmowego i że centralna aktywacja receptorów kannabinoidowych jest konieczna do pełnego procesu gojenia. Oś jelitowo-mózgowa również może odgrywać ważną rolę w tym procesie.
Inną opcją minimalizacji niepożądanych skutków psychotropowych podczas leczenia zapalenia przewodu pokarmowego może być wybór niższej dawki kannabinoidów. Z badań przedklinicznych wiadomo, że stosowanie niskich dawek THC poprawia działanie np. na miażdżycę [ 86 ] i funkcje poznawcze [ 87 ]. U pacjentów z przewlekłym zespołem górnego neuronu ruchowego leczenie nabilonem w dawce 1 mg/dzień znacząco zmniejszyło ból [ 88 ]. Co ważne, w tym badaniu nabilon był dobrze tolerowany. Badanie kliniczne przeprowadzone na starszych pacjentach z demencją wykazało również, że niskie dawki THC (0,75 mg dwa razy dziennie) są bezpieczne i dobrze tolerowane; nie było różnic w odczuwaniu „haju” pomiędzy grupą otrzymującą lek i grupą placebo [ 89 ]. Ze względu na znaczącą ekspresję CB 1 w mózgu, jelitowym układzie nerwowym i nabłonku jelit, leczenie małymi dawkami kannabinoidów w IBD może aktywować receptory kannabinoidowe zarówno ośrodkowo, jak i obwodowo, przyczyniając się w ten sposób do gojenia się ran, przywrócenia funkcji barierowych i rozluźnienia jelit.
Wreszcie, inhibitory enzymów degradujących EC podnoszą poziomy EC i są odpowiednie do leczenia zapalenia przewodu pokarmowego. Na podstawie wyników badań przedklinicznych można stwierdzić, że inhibitory MGL mogą być obiecującymi kandydatami do leczenia IBD [ 32 ], ale również inhibitory FAAH okazały się skuteczne w łagodzeniu eksperymentalnego zapalenia jelit [ 35 ]. Jednakże przełożenie tych skutków na choroby przewodu pokarmowego u ludzi jest opóźnione, szczególnie w przypadku inhibitorów FAAH, ponieważ badania kliniczne zostały wstrzymane ze względu na incydent z BIA 10-2474 [ 90 ]. Ponadto możliwość wystąpienia sercowo-naczyniowych skutków ubocznych inhibitorów FAAH może również ograniczać ich stosowanie u ludzi [ 91 , 92 , 93 ].
Konopie indyjskie /kannabinoidy w CRC
Korzystne działanie kannabinoidów na CRC znane jest jedynie z eksperymentów na myszach. Nokaut CB 1 u myszy Apc Min/+ i C57BL/6 [ 20 , 21 ] powoduje, w zależności od zastosowanego modelu, zwiększone obciążenie nowotworem w jelicie cienkim i okrężnicy, co wskazuje na supresorową rolę CB 1 w CRC. Natomiast CB 2 powiązano z postępem CRC [ 22 , 94 ], co utrudnia ustalenie jasnego uzasadnienia leczenia kannabinoidami u ludzi CRC. Niezbędnych jest zatem więcej danych przedklinicznych na temat roli CB 2 w doświadczalnym CRC, aby stworzyć podstawę do przełożenia wyników eksperymentalnych na praktykę kliniczną. Ponieważ kannabinoidy zostały zatwierdzone do leczenia wymiotów i nudności u pacjentów chorych na nowotwory poddawanych chemioterapii, z obserwacji tych pacjentów możemy dowiedzieć się, czy przeciwwymiotne leczenie kannabinoidami wpływa na progresję nowotworu podczas chemioterapii. W tym kontekście należy wspomnieć, że połączenie THC z CBD skutecznie zwiększa śmierć komórek i migrację linii komórkowych szpiczaka mnogiego w synergii z karfilzomibem, inhibitorem proteasomu, stosowanym w leczeniu szpiczaka mnogiego [ 95 ]. Temat terapii kannabinoidami w CRC został również poruszony w niedawnym artykule redakcyjnym [ 63 ].
Wniosek
Eksperymentalne modele zapalenia jelit pokazują, że pomocne może być leczenie kannabinoidami. Również w modelach eksperymentalnych CRC wykazano , że CB 1 ma działanie ochronne. Jednakże jeśli chodzi o mechanizmy działania, wiadomo, że kannabinoidy mogą przekazywać sygnał wieloma szlakami, najprawdopodobniej w sposób oparty na ligandach [ 96 ]. Pomimo przytłaczającej ilości danych wskazujących, że kannabinoidy mają właściwości przeciwnowotworowe in vitro, dane in vivo potwierdzające ich działanie są nieliczne. Niektóre receptory reagujące na kannabinoidy, takie jak GPR55, wykazały nawet właściwości prozapalne i prokarcynogenne. Zatem ze względu na złożoność mechanizmów w obrębie ECS, korzystny wpływ fitokannabinoidów i syntetycznych kannabinoidów na ludzi nie zawsze jest pewny. Ponadto psychotropowe działanie THC i syntetycznych agonistów CB 1 w dalszym ciągu utrudnia ich szersze zastosowanie, czego można uniknąć poprzez strategie takie jak stosowanie kannabinoidów w małych dawkach z minimalnymi efektami ośrodkowymi lub stosowanie ligandów receptorów kannabinoidowych działających obwodowo. Każde podejście ma wady i wiele pytań. Z pewnością potrzebne są duże badania kliniczne, aby stworzyć solidne wytyczne dotyczące bezpieczeństwa, skuteczności, dawkowania, drogi, formy stosowania i skutków ubocznych kannabinoidów. Mamy nadzieję, że w najbliższej przyszłości dane kliniczne zapewnią podstawę do tego, czy konopie indyjskie lub kannabinoidy mogą stać się wartościowymi lekami, przynajmniej w leczeniu IBD. W przypadku CRC zajmie to z pewnością więcej czasu.
Rak jelita grubego jest poważnym problemem zdrowia publicznego. Niestety, obecnie nie istnieje żadna skuteczna opcja leczenia tego typu nowotworu złośliwego. Najbardziej obiecującym obecnie leczeniem raka jest immunoterapia, która jest również nazywana terapią biologiczną lub ukierunkowaną. Ten rodzaj terapii wzmacnia zdolność układu odpornościowego pacjenta do zwalczania nowotworu złośliwego. Jednak komórki nowotworowe mogą stać się oporne na immunoterapię i uniknąć nadzoru immunologicznego poprzez uzyskanie zmian genetycznych. Dlatego też konieczne są nowe strategie leczenia. W ostatniej dekadzie kilka raportów sugeruje skuteczność kannabinoidów i ekstraktów z Cannabis sativa w hamowaniu proliferacji nowotworów in vitro i in vivo , w tym nowotworów złośliwych jelit. Wykazano, że kannabinoidy modulują ścieżki zaangażowane w proliferację komórek, angiogenezę, zaprogramowaną śmierć komórek i przerzuty. Z tego powodu są proponowane jako terapia wspomagająca w przypadku wielu nowotworów złośliwych. O wiele mniej informacji istnieje na temat potencjału stosowania konopi w połączeniu z immunoterapią. W tym artykule analizujemy możliwość wykorzystania kannabinoidów w modulacji immunoterapii raka jelita grubego i omawiamy możliwe zalety i ograniczenia.
Obecnie rak jelita grubego (CRC) jest uważany za trzeci najbardziej śmiertelny i czwarty najczęściej wykrywany nowotwór na świecie ( 1 ). Pomimo obecności wysoce zaawansowanych technik przesiewowych, wskaźnik zapadalności stale rośnie na całym świecie ( 2 ). Szacuje się, że globalne obciążenie rakiem jelita grubego wzrośnie o 60% do ponad 2,2 miliona nowo zdiagnozowanych przypadków i 1,1 miliona zgonów do 2030 roku ( 3 ). Czynniki takie jak siedzący tryb życia, zwiększone spożycie alkoholu, tytoniu, czerwonego mięsa, predyspozycje genetyczne, przewlekłe procesy zapalne przewodu pokarmowego są czynnikami wyzwalającymi tego typu złośliwość ( 4 ). Wiadomo, że głównymi prekursorami CRC są polipy gruczolakowate. Szybkość transformacji tych polipów w raka wynosi ~0,25% rocznie. Gdy te zmiany mają wysoki stopień dysplazji i architekturę kosmków, ryzyko transformacji w nowotwór złośliwy wzrasta do 50% ( 5 ).
Zrozumienie patogenezy raka jelita grubego jest bardzo ważne dla wyboru właściwej terapii. Etiologia CRC jest złożona i obejmuje akumulację nabytych modyfikacji epigenetycznych i genetycznych, które przekształcają normalne komórki nabłonkowe w złośliwe. Klasyczny model progresji nowotworu nazywa się rozwojem sekwencji polipa-raka, która obejmuje trzy główne etapy. Pierwszym etapem jest tworzenie łagodnych nowotworów, takich jak gruczolaki i siedzące ząbkowane polipy. Drugi etap charakteryzuje się progresją łagodnych nowotworów w nowotwory bardziej zaawansowane histologicznie, a ostatni etap – ich transformacją w raka. Proces ten może trwać wiele lat bez wykazywania jakichkolwiek oznak i objawów. Kiedy CRC się rozwinie, może minąć jeszcze kilka lat, zanim zostanie zdiagnozowany. CRC jest spowodowany mutacjami w onkogenach, genach supresorowych nowotworu i genach zaangażowanych w mechanizmy naprawy DNA. Jedna z pierwszych mutacji występuje zazwyczaj w genie polipowatości gruczolakowatej jelita grubego (APC), który jest supresorem nowotworu, po czym następują mutacje w genach KRAS, TGF-β, BAX, BRAF i innych ( 6 ).
Większość przypadków CRC jest sporadyczna (70–80%), podczas gdy dziedziczne i rodzinne przypadki CRC stanowią odpowiednio około 5 i 25%. Sporadyczne nowotwory powstają z powodu mutacji punktowych, a molekularna patogeneza tych nowotworów jest bardzo niejednorodna. Dziedziczna grupa tego konkretnego nowotworu złośliwego jest spowodowana odziedziczonymi mutacjami i może być podzielona na dwie grupy: polipowatość i niepolipowatość. Typ polipowatości obejmuje głównie rodzinną polipowatość gruczolakowatą, która charakteryzuje się obecnością licznych, prawdopodobnie złośliwych polipów w jelicie grubym. Wariant niepolipowatości jest reprezentowany przez zespół Lyncha ( 7 ). Rodzinny CRC jest również spowodowany odziedziczonymi mutacjami i występuje rodzinnie bez obecności konkretnych dziedzicznych zespołów ( 8 ).
Ostatnio zaproponowano dwie molekularne klasyfikacje patologiczne oparte na szeroko zakrojonej analizie genomicznej i transkryptomowej CRC. Pierwsza z nich nazywa się The Cancer Genome Atlas (TCGA) i obejmuje trzy grupy: hipermutacje (13%), ultramutacje (3%) i niestabilność chromosomową (84%). Kategoria hipermutacji charakteryzuje się wysokim wskaźnikiem mutacji, wadliwą naprawą niedopasowań (dMMR) z dobrym rokowaniem, ale złym rokowaniem po nawrocie. Typ ultramutacji ma niezwykle wysoki wskaźnik mutacji z mutacją korekty epsilon polimerazy DNA i ogólnie dobrym rokowaniem. Większość CRC wyróżnia się niestabilnością chromosomową (CIN) z cechami niskiej częstości mutacji, ale wysoką częstością zmian liczby kopii somatycznych DNA. Druga klasyfikacja oparta na ekspresji genów nazywa się Consensus Molecular Subtypes (CMS) i obejmuje cztery grupy. CMS1 (14%) charakteryzuje się niestabilnością mikrosatelitarną (MSI), mutacją onkogenu BRAF i silną aktywacją immunologiczną. U pacjentów z tym podtypem zauważono słaby wskaźnik przeżycia po nawrocie. CMS2 (37%), zwany również kanonicznym, wykazuje wysoką niestabilność chromosomową i aktywację sygnalizacji WNT i MYC. CMS3 (13%), znany jako metaboliczny, ma liczne mutacje KRAS i deregulowane ścieżki metabolizmu. CMS4 (23%), zwany mezenchymalnym, jest opisany obecnością nacieku podścieliska, silnie ekspresjonowanymi genami mezenchymalnymi, aktywacją transformującego czynnika wzrostu beta, gorszym ogólnym i wolnym od nawrotów przeżyciem w porównaniu z pacjentami z innych grup ( 7 , 9 ). Klasyfikacje te dostarczyły informacji na temat właściwego doboru leczenia i rokowania pacjentów, co jest bardzo ważne dla trwających i przyszłych badań klinicznych.
Główne opcje terapeutyczne dostępne obecnie dla pacjentów z CRC to chirurgia, chemioterapia, immunoterapia, radioterapia. 5-letni wskaźnik przeżycia pacjentów z wczesnymi stadiami CRC wynosi prawie 90%. Ze względu na subtelne objawy, u ponad połowy pacjentów diagnozuje się chorobę, gdy rozwinęły się już zaawansowane nowotwory złośliwe. 5-letni wskaźnik przeżycia wynosi tylko 10% lub mniej, gdy pacjenci mają przerzuty ( 10 ).
Wśród nowych potencjalnych podejść terapeutycznych wykazano, że leczenie kannabinoidami i ekstraktami z Cannabis sativa jest skuteczne w hamowaniu wzrostu raka in vitro i in vivo ( 11 ). Roślina C. sativa zawiera fitokannabinoidy, terpenoidy, flawonoidy, kwasy tłuszczowe i inne cząsteczki. Kannabinoidy działają poprzez układ endokannabinoidowy, który składa się z receptorów, takich jak kannabinoid 1 (CB1), kannabinoid 2 (CB2), kanały potencjału receptora przejściowego podtypu waniloidowego 1 i 2 (TRPV1, TRPV2), receptory sprzężone z białkiem G 18, 55, 119 (GPR18, GPR55, GPR119), endokannabinoidy, takie jak 2-arachidonoiloglicerol i anandamid (2-AG, AEA) oraz enzymy odpowiedzialne za ich metabolizm. Głównymi enzymami biosyntetycznymi są NAPE-fosfolipaza D (NAPE-PLD) i lipaza diacyloglicerolowa (DAGL); głównymi enzymami degradacji są hydrolaza amidu kwasu tłuszczowego (FAAH) i lipaza monoacyloglicerolowa (MAGL). Główną funkcją układu endokannabinoidowego jest utrzymanie homeostazy ( 12 ). Receptor CB1 jest głównie ekspresowany w ośrodkowym układzie nerwowym, a receptor CB2, będący najbardziej rozpowszechnionym w układzie odpornościowym, występuje głównie w narządach obwodowych. Oba receptory są receptorami powierzchniowymi komórek sprzężonymi z białkiem G, które są sprzężone ze szlakami cyklazy adenylowej i kinazy białkowej cAMP A oraz szlakami MAPK i PI3K ( 13 ).
Znaczenie układu odpornościowego w raku jelita grubego
W przeszłości guzy były definiowane jako zbiór jednorodnych komórek nowotworowych. Agresywność nowotworu opisywano na podstawie jego cech kliniczno-patologicznych. Ostatnie postępy w immunologii i biologii molekularnej pozwoliły nam lepiej poznać podstawowe mechanizmy potencjału przerzutowego guzów. Wiele badań w tej dziedzinie poszerzyło wiedzę i podkreśliło znaczenie układu odpornościowego w regulacji wzrostu nowotworu. Głównymi graczami tego procesu są wrodzone komórki odpornościowe, takie jak neutrofile, makrofagi, komórki tuczne, eozynofile, komórki supresorowe pochodzące z mieloidu (MDSC) oraz adaptacyjne komórki odpornościowe, takie jak limfocyty T i B ( 14 , 15 ).
W ciągu ostatniej dekady wiedza na temat mikrośrodowiska guza (TME) stała się kluczem do zrozumienia złożonej, wieloetapowej tumorigenezy i opracowania nowych schematów leczenia i leków ( 16 ). Mikrośrodowisko raka obejmuje komórki rezydentne i nierezydentne, które są połączone ze sobą różnymi mediatorami, a każda z nich ma określoną funkcję. Komunikacja między tymi komórkami a komórkami nowotworowymi w ich otoczeniu zasadniczo reguluje los progresji guza. Komórki odpornościowe mogą hamować lub sprzyjać wzrostowi guza ( Tabela 1 ). Nowe badania przedkliniczne wykazały, że atypowe komórki nieprezentujące antygenu są najpierw atakowane przez wrodzony układ odpornościowy; następnie odpowiedź zapalna promuje tworzenie nowych naczyń krwionośnych i proliferację komórek nowotworowych. Niestety, guzy mogą włączać mechanizmy immunosupresyjne i unikać immunonadzoru gospodarza. Adaptacyjna odpowiedź immunologiczna wymaga identyfikacji antygenów nieswoistych poprzez komunikację między białkami a głównym kompleksem zgodności tkankowej komórek prezentujących antygen i receptorami komórek CD8+ i CD4+ T poprzez prezentację antygenu. Guzy mogą utracić swoją antygenowość z powodu nabytych wad w prezentacji antygenu lub mogą zostać zidentyfikowane jako własne ( 25 – 27 ).
Tabela 1.
Działanie pronowotworowe i przeciwnowotworowe komórek układu odpornościowego.
Komórki odpornościowe
Rola w nowotworach (przeciwnowotworowa i pronowotworowa)
Odniesienia
Komórki dendrytyczne (DC)
Uwolnij cytotoksyczne cytokiny Prezentacja antygenu limfocytom T
Wspomaganie wzrostu nowotworu poprzez stymulację neoangiogenezy, przebudowę tkanek i modulację odpowiedzi immunologicznej gospodarza
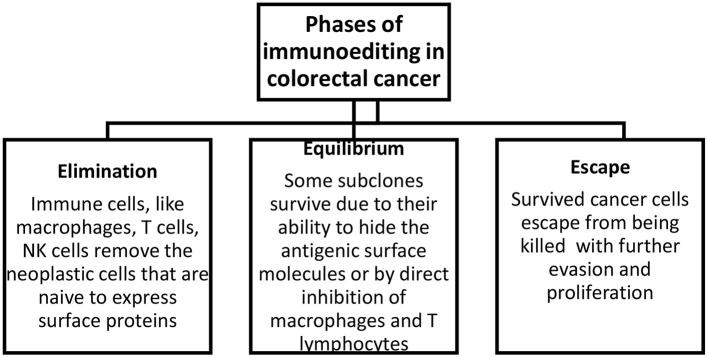
Istnieją trzy fazy immunoedycji guza: eliminacja, równowaga i ucieczka ( Rysunek 1 ). W pierwszej fazie komórki układu odpornościowego eliminują komórki nowotworowe, które ekspresują białka powierzchniowe. W fazie równowagi niektóre komórki utrzymują się dzięki swojemu potencjałowi do kamuflowania cząsteczek powierzchniowych lub poprzez tłumienie makrofagów i komórek T poprzez ekspresję substancji takich jak PD-1/2 na komórkach prezentujących antygen. W ostatniej fazie niektóre komórki mogą uniknąć zabicia, co następnie prowadzi do ucieczki i proliferacji opornych klonów. Ponadto degradacja macierzy zewnątrzkomórkowej przez metaloproteinazy macierzy i nowe naczynia krwionośne utworzone w wyniku nieprawidłowej angiogenezy sprzyjają tworzeniu się przerzutów ( 15 ).
Rysunek 1.
Fazy immunoedycji w raku jelita grubego. Eliminacja obejmuje usunięcie komórek nowotworowych, równowaga opisuje przeżycie frakcji przekształconych komórek, a ucieczka opisuje ucieczkę i proliferację tych komórek.
Jeśli chodzi o ekspresję receptorów kannabinoidowych w komórkach układu odpornościowego, wykazano, że receptory te są wyrażane zarówno w odporności adaptacyjnej, jak i wrodzonej. Na przykład receptory CB1, CB2 i GPR55 są wyrażane na komórkach NK, CB1, CB2 – na komórkach tucznych, limfocytach T – na komórkach B. Dlatego można postawić hipotezę, że fitokannabinoidy mogą wpływać na funkcjonowanie układu odpornościowego, regulować stan zapalny i posiadać działanie przeciwnowotworowe itp. ( 28 ).
Rola stanu zapalnego w karcynogenezie jelita grubego
Zapalenie odgrywa kluczową rolę w karcynogenezie jelita grubego i jest obecnie uważane za jedną z pojawiających się cech charakterystycznych raka ( 29 ). Lepsze zrozumienie CRC i zapalenia może doprowadzić do opracowania nowych biomarkerów nowotworowych i bardziej spersonalizowanych i skutecznych terapii. Wiadomo, że pacjenci cierpiący na przewlekłe schorzenia, takie jak choroba zapalna jelit, mają znacznie wyższe ryzyko rozwoju CRC ( 30 ). Zapalenie jest uważane za ważną siłę napędową nowotworów CRC związanych z zapaleniem jelita grubego, podczas gdy jego rola w przypadku nowotworów sporadycznych i dziedzicznych jest mniej jasna. Dowody wskazują, że niesteroidowe leki przeciwzapalne mogą zapobiegać rozwojowi CRC lub go opóźniać ( 31 ). Metaanaliza badań z randomizacją wykazała, że podczas obserwacji po 20 latach stosowania aspiryny przez 5 lat śmiertelność i częstość występowania CRC uległyby zmniejszeniu o 30–40% ( 32 ).
Na podstawie klasyfikacji CMS CRC, CMS1 i CMS4 są uważane za zapalne, przy czym pierwszy ma złe rokowanie po nawrocie, a drugi — najgorszy wskaźnik przeżycia. Ogólnie rzecz biorąc, stan zapalny odgrywa podwójną rolę w nowotworze. Celowanie w komórki złośliwe przez cytotoksyczne limfocyty T lub zmniejszanie niespecyficznego stanu zapalnego przez T-regs może prowadzić do odpowiedzi przeciwnowotworowej. Ten typ odpowiedzi nazywa się ochronną i jest związany z polaryzacją Th1 i niższym nawrotem CRC. Podtyp Th1 wytwarza IFN-γ i wzmacnia toksyczność komórkową, podczas gdy podtyp Th2 uwalnia IL-4 i wzmacnia humoralną odpowiedź komórek B. Najczęstszymi cytokinami prozapalnymi są TNF-α, IL-6, IL-12, IL-2, a najczęstszymi przeciwzapalnymi są IL-10, IL-4, IL-5, TGF-β i IFN-α ( Tabela 2 ). Komórki odporności wrodzonej i nabytej oraz inne komórki, takie jak fibroblasty, komórki mezenchymalne i perycyty, odgrywają ważną rolę w zapaleniu związanym z rakiem ( 57 ). Komunikacja między tymi komórkami odbywa się za pośrednictwem sieci cytokin produkowanych i wydzielanych przez komórki układu odpornościowego po stymulacji. Rola szlaku sygnałowego IL-10 pozostaje kontrowersyjna w przypadku CRC. Wyższy poziom IL-10 wiąże się z gorszym przeżyciem pacjentów, podczas gdy badania na zwierzętach pokazują, że pełni on rolę ochronną poprzez tłumienie stanu zapalnego ( 46 , 47 ). IL-6 jest aktywatorem szlaku sygnałowego STAT-3 i często występuje u pacjentów z CRC; i jest również powiązany z gorszym przeżyciem i zwiększonym ryzykiem nawrotu ( 37 , 57 , 58 ). Fibroblasty podścieliska, uzyskane z raka jelita grubego, wyprodukowały znaczne ilości IL-6. Ostatni z nich indukował angiogenezę guza poprzez zwiększenie produkcji VEGF ( 38 ). IL-6 ułatwia kolonizację przerzutową komórek raka jelita grubego. U myszy IL6-/- przerzuty komórek CT26 do wątroby zostały zmniejszone, a funkcja komórek CD8+ T uległa poprawie in vivo . Co więcej, myszy z niedoborem IL-6 skutecznie odpowiedziały na wstrzyknięcie anty-PD-L1 poprzez zahamowanie kolonizacji przerzutowej, podczas gdy u myszy IL6+/+ nie zaobserwowano tego efektu ( 39 ). IFN-γ jest wytwarzany przez komórki CD4+, CD8+ i NK i indukuje apoptozę komórek. Utrata jednej kopii tego interferonu u myszy Apcmin/+ wykazała znacznie szybszy postęp w kierunku gruczolakoraka jelita grubego. Komórki CRC mogą minimalizować przeciwnowotworowe efekty sygnalizacji interferonu przez łańcuch receptora interferonu typu I, co prowadzi do słabej odpowiedzi na inhibitory punktów kontrolnych anty-PD1 ( 44 ). Ekspresja TNF-α jest znacznie wyższa w raku jelita grubego niż w sąsiadującej prawidłowej tkance jelita grubego. Zwiększona ekspresja tej cytokiny silnie koreluje z bardziej zaawansowanymi nowotworami ( 59). Po stymulacji TNF-α zauważono wzrost onkogenu Metastasis-Associated in Colon Cancer 1 (MACC1) zarówno na poziomie mRNA, jak i białka. MACC1 indukuje proliferację, przeżycie i przerzuty komórek nowotworowych. Poziomy ekspresji tego onkogenu zostały zmniejszone przez obniżenie p65 NF-kB. Ponadto indukcję MACC1 utrudniało monoklonalne przeciwciało anty-TNF-α, adalimumab ( 34 ). TNF-α zwiększał poziomy cytokin prozapalnych, takich jak IL-6 i IL-8 in vitro na komórkach raka jelita grubego HT-29 ( 35 ). Inne badanie wykazało, że wpływ szczepionki peptydowej, AH1, na myszy z guzem jelita grubego CT26 spowodował skromne zahamowanie wzrostu guza, ale połączenie z F8-TNF drastycznie zwiększyło aktywność przeciwnowotworową. F8-TNF to białko fuzyjne przeciwciał, które dostarcza TNF do macierzy zewnątrzkomórkowej guza. Synergia między szczepionką peptydową a białkiem fuzyjnym TNF została wyjaśniona tym, że F8-TNF powoduje szybką martwicę krwotoczną guza i w rezultacie pozostawia niewielką ilość resztkowych komórek nowotworowych. Ponadto zauważono znaczący wzrost specyficznych dla AH1 komórek CD8+ T w guzach i drenujących węzłach chłonnych ( 60 ). IL-12 hamowała ruchliwość i inwazję ludzkich komórek raka jelita grubego (HRT18, HT29 i HT115), co sugeruje jej ważną rolę w przerzutach ( 41 ). IL-4 jest aktywnie uwalniana przez komórki macierzyste raka jelita grubego i nadaje guzom fenotyp odporny na śmierć. Neutralizacja IL-4 za pomocą przeciwciała znacząco uwrażliwia komórki nowotworowe na chemioterapię ( 49 ). Wczesna transgeneza IL-5 w mysim modelu CRC z zapaleniem jelita grubego zwiększyła ciężkość zapalenia jelita grubego, wywołała szybkość tworzenia polipów i w rezultacie większe obciążenie guzem ( 51 ). U pacjenta zgłoszono przypadek skrajnej eozynofilii spowodowanej przez rozsianego raka jelita grubego wytwarzającego IL-5 ( 61 ). TGF-β promuje przeżycie, inwazję i przerzuty komórek CRC ( 53 ). TGB-β w mikrośrodowisku guza zwiększa wykluczenie komórek T i hamuje wykopywanie fenotypu Th-1. Myszy z przerzutowym rakiem jelita grubego i zablokowaną ścieżką sygnałową TGF-β mają guzy wrażliwe na terapię anty-PD-1 anty-PD-L1. Natomiast myszy z odblokowaną sygnalizacją TGF-β wykazały ograniczoną odpowiedź na inhibitory punktów kontrolnych układu odpornościowego ( 54 ). Systemowe podanie IFN-α myszom z rakiem jelita grubego znacząco zahamowało wzrost guza i jego unaczynienie; indukowanej apoptozy komórek nowotworowych i komórek śródbłonka wątroby związanych z przerzutami ( 56 ).
Tabela 2.
Główne efekty cytokin pro- i przeciwzapalnych.
Cytokiny
Miejsce produkcji
Ruchomości
Znaczenie dla karcynogenezy jelita grubego
Odniesienia
Cytokiny prozapalne
TNF-alfa
Makrofagi, limfocyty T, komórki NK, komórki tuczne, eozynofile
Stymulacja stanów zapalnych, odporność na infekcje i nowotwory
TNF-α reguluje indukcję MACC1 poprzez podjednostkę NF-κB p65. Zwiększa poziom IL-6 i IL-8.
Optymalny sposób leczenia można wybrać, łącząc i analizując informacje na temat czynników związanych z nowotworem (lokalizacja nowotworu, obecność przerzutów, obecność biomarkerów itp.) oraz czynników związanych z pacjentem (rokowanie, choroby współistniejące itp.).
Pacjenci z rakiem jelita grubego z chorobą przerzutową otrzymują kombinację chemioterapii i immunoterapii. Chemioterapia pierwszego rzutu obejmuje fluoropirymidyny, takie jak kapecytabina i 5-fluorouracyl (5-FU) w monoterapii lub z leukoworyną (LV), oksaliplatyną (5-FU/LV/oksaliplatyna – FOLFOX), irynotekanem (5-FU/LV/irynotekan – FOLFIRI), kapecytabiną/LV/oksaliplatyną – CAPOX. Chemioterapia drugiego rzutu – FOLFOX lub CAPOX dla pacjentów opornych na irynotekan. Pacjentom opornym na kombinacje oksaliplatyny przepisuje się FOLFIRI lub irynotekan w monoterapii. Zwykle leczenie trwa do 6 miesięcy, ale czas trwania w znacznym stopniu zależy od indywidualnych przypadków ( 62 ).
Najczęstsze działania niepożądane chemioterapii w przypadku CRC to leukopenia, polineuropatia, biegunka, trombocytopenia, nadmierne wymioty, dysfunkcje wątrobowo-nerkowe i pogorszenie ogólnego stanu. Nasilenie działań niepożądanych jest zwykle większe u pacjentów w podeszłym wieku i u pacjentów z wcześniej istniejącymi chorobami współistniejącymi ( 63 ). Ze względu na obawy dotyczące toksyczności chemioterapia może nie być odpowiednia dla wielu pacjentów. Onkolodzy mogą nie zalecać tego typu leczenia ze względu na niektóre zaawansowane stadia chorób przewlekłych (niewydolność wątroby, nerek i serca) i słabą sprawność fizyczną ( 64 ).
Immunoterapia
Immunoterapia jest jedną z najbardziej obiecujących metod terapeutycznych dla pacjentów z rakiem jelita grubego ( 65 ). Terapia celowana zrewolucjonizowała leczenie raka. Immunoterapia jest rodzajem podejścia leczniczego, które pomaga układowi odpornościowemu w eliminacji guzów. Można ją podzielić na dwie główne grupy: aktywną (szczepionki) i pasywną (przeciwciała monoklonalne, adoptywna terapia komórkowa) ( Tabela 3 ). Ponadto niektóre terapie biologiczne mogą być szczególnie ukierunkowane na określone antygeny nowotworowe, podczas gdy inne działają niespecyficznie, wzmacniając naturalne odpowiedzi immunologiczne ( 75 ).
Tabela 3.
Zalety i wady środków immunoterapeutycznych.
Immunoterapia
Zalety i wady
Status zatwierdzenia w CRC
Odniesienia
Szczepionki na cały guz
Składa się ze wszystkich znanych i nieznanych antygenów nowotworowych, łatwa produkcja
Istnieją pewne rodzaje szczepionek przeciwnowotworowych, które badano w leczeniu CRC, takie jak szczepionki na cały guz, peptydy, wektory wirusowe i komórki dendrytyczne (DC). Celem tych środków, podobnie jak każdej innej strategii immunizacji, jest wywołanie odpowiedzi immunologicznej przeciwnowotworowej, która wyeliminuje raka i zapewni organizmowi ciągły nadzór w celu ochrony przed jego nawrotem.
Szczepionki na cały guz
Niektóre zalety pracy ze szczepionkami na cały guz to: są łatwe w produkcji i składają się ze wszystkich znanych i nieznanych antygenów nowotworowych. Natomiast najistotniejszą wadą tych szczepionek jest bardzo niska immunogenność, która może być skierowana do normalnych komórek, a w rezultacie niska skuteczność. Podjęto kilka podejść w celu zwiększenia immunogenności szczepionek na cały guz. Przeprowadzono badanie z zakażeniem wirusem choroby Newcastle, które wykazało 98% 2-letni wskaźnik przeżycia u pacjentów z wyciętym rakiem jelita grubego w porównaniu z 67% u pacjentów, którzy otrzymali szczepionkę na cały guz w połączeniu ze szczepionką Bacillus Calmette-Guérin (BCG). Wyniki sugerują, że immunogenność tych związków została poprawiona ( 27 ).
Szczepionki peptydowe
Szczepionki peptydowe są bardziej specyficzne dla antygenu związanego z guzem, ale ich skuteczność jest nadal znacznie niska ze względu na niewielką ilość odpowiedzi komórek T. Badanie fazy I/II przeprowadzone u pacjentów z rakiem jelita grubego wykazało, że połączenie szczepionki p53 z interferonem alfa zwiększyło ilość interferonu gamma ( 76 ). Następny typ szczepionki to szczepionki wektorowe wirusowe. Są specyficzne dla antygenu związanego z guzem i naturalnie immunogenne. Wadą ich stosowania jest ich zdolność do wywoływania burzy cytokinowej. Najczęściej stosowanymi wirusami w raku jelita grubego są adenowirusy, pokswirusy i alfawirusy. Większość z tych szczepionek jest ukierunkowana na antygen karcinoembrionalny (CEA), białko wyrażane przez raki jelita grubego. Dane przedkliniczne pokazują, że rekombinowany wirus ospy wyrażający CEA (rV-CEA) może wzmacniać adaptacyjne i wrodzone odpowiedzi immunologiczne u myszy. Ponadto hamował proliferację gruczolakoraka jelita grubego w modelach zwierzęcych. Jednakże badania kliniczne przeprowadzone na pacjentach w zaawansowanym stadium raka jelita grubego wykazały brak odpowiedzi ( 77 ). Szczepionki z komórek dendrytycznych charakteryzują się specyficznością antygenu związanego z guzem i generowaniem własnej odpowiedzi immunologicznej organizmu. Negatywnymi aspektami są wysokie koszty i bardzo czasochłonny proces przygotowania. Po całkowitym wycięciu przerzutów CRC do wątroby, badanie kliniczne szczepionki fazy II wykazało mniej i opóźnione nawroty w grupie szczepionej w porównaniu z grupą obserwacyjną ( 78 ). Wyniki szczepionek DC są bardzo zachęcające, a wkrótce ich skuteczność może zostać znacznie poprawiona.
Terapia Adopcyjnego Transferu Komórek
Adopcyjna terapia transferu komórek to kolejny rodzaj immunoterapii. Głównymi zaletami tego typu leczenia są eliminacja konieczności wytworzenia odpowiedzi immunologicznej i wysoka swoistość guza. Z kolei pewnymi wadami są wysokie koszty, długi czas przygotowania i toksyczność zależna od celu. W tej terapii autologiczne komórki T są pobierane z guza, węzłów chłonnych lub krwi obwodowej pacjenta i modyfikowane ex vivo poprzez ich ekspansję i dodanie niektórych cząsteczek współstymulujących i cytokin. Następnie przeprowadza się bierny transfer tych komórek T do gospodarza w celu bezpośredniego zniszczenia guza. Najnowszym odkryciem tego typu biernej immunoterapii jest opracowanie zmodyfikowanych komórek T, które wyrażają chimeryczne receptory antygenowe specyficznie dla antygenu karcinoembrionalnego ( 79 ). Badanie fazy I przeprowadzone u pacjentów z rakiem jelita grubego opornym na standardowy schemat protokołu leczenia przy użyciu autologicznych komórek T zmodyfikowanych w celu wyrażenia receptora komórek T CEA myszy wykazało znaczący spadek stężenia CEA w surowicy u wszystkich trzech pacjentów; u jednego z nich wykazano odpowiedź kliniczną w postaci regresji przerzutów w wątrobie i płucach. Jednocześnie u tych pacjentów wystąpiło przejściowe zapalenie jelit ( 80 ). Niedawno opublikowane wyniki badania przypadku u pacjentów z zaawansowanym rakiem jelita grubego wykazały znaczącą odpowiedź kliniczną na połączenie kapecytabiny i adopcyjnego transferu komórek (komórek αβT i komórek NK) przepisanych po laparoskopowej resekcji raka jelita grubego i niektórych przerzutów do wątroby. Dwa tygodnie po laparoskopii zaobserwowano drastyczny wzrost poziomu CEA. Adopcyjny transfer komórek pozwolił na obniżenie poziomu CEA w surowicy, ostatecznie doprowadzając go do normy. Zaobserwowano zauważalną redukcję wielkości nieoperacyjnych przerzutów do wątroby. Podczas badania kontrolnego po 19 miesiącach nie odnotowano progresji ani nawrotu, a poziomy CEA utrzymywały się w granicach normy ( 81 ).
Immunoterapia oparta na przeciwciałach
Wysoce specyficzne przeciwciała monoklonalne są bardzo skuteczne w leczeniu raka od dziesięcioleci. Białka przeciwko receptorowi naskórkowego czynnika wzrostu (EGFR) i czynnikowi wzrostu śródbłonka naczyniowego (VEGF) w połączeniu z chemioterapią wykazały lepsze wyniki leczenia złośliwego CRC (mCRC). Środki anty-EGFR, takie jak Cetuximab lub Panitumumab, w monoterapii lub w połączeniu z lekami cytotoksycznymi są przepisywane tylko wtedy, gdy nie ma mutacji KRAS ( 62 ). Bevacizumab, humanizowane przeciwciało monoklonalne przeciwko VEGF, hamuje wzrost guza i angiogenezę, a także moduluje układ odpornościowy gospodarza poprzez zwiększenie populacji komórek B i T ( 82 ).
Dramatyczną skuteczność immunoterapii opartej na przeciwciałach udowodniono przy użyciu innego typu przeciwciał monoklonalnych (mAbs) znanych jako inhibitory punktów kontrolnych (ICI). Wykazano, że obecnie stosowane ICI zapewniają znaczące odpowiedzi kliniczne u pacjentów z mCRC, szczególnie z typem niedoboru naprawy niedopasowania/wysokiej niestabilności mikrosatelitarnej (dMMR-MSI-H). Celują one w hamujące receptory immunologiczne: programowaną śmierć komórki 1 (PD-1) i jej ligand PD-L1, cytotoksyczny antygen związany z limfocytami T 4 (CTLA-4). Ten ostatni jest wyrażany w naiwnych limfocytach T, efektorowych limfocytach T i regulatorowych limfocytach T (T-regs). Stymuluje on dezaktywację T-regs poprzez wiązanie się z komórkami prezentującymi antygen. Receptor PD-1 jest obecny w limfocytach CD4, CD8, komórkach NK, MDSC, T-regs i limfocytach B. Razem ze swoim ligandem, ten receptor powoduje wyczerpanie komórek T poprzez minimalizowanie naciekających guz limfocytów i proliferacji komórek T. W konsekwencji, guzy nabywają immunooporności. Środki anty-CTLA-4 i anty-PD-1 aktywują komórki T i powodują silniejszą odpowiedź przeciwnowotworową ( 83 ). Guzy CRC dMMR-MSI-H mają 20 razy większy ładunek mutacji niż guzy z niestabilnością mikrosatelitarną o niskiej sprawności naprawy niezgodności (pMMR-MSI-L). Ponadto są bardziej naciekane przez TIL, makrofagi i mają podwyższone poziomy cytokin stymulujących układ odpornościowy w porównaniu z pMMR-MSI-L. Ten ostatni ma mniej skuteczną odpowiedź na ICI i gorsze rokowanie ( 84 ).
W sierpniu 2020 r. istniały trzy zatwierdzone przez FDA ICI, które były stosowane u pacjentów z dMMR-MSI-H mCRC. Pierwszym z nich był Nivolumab, lek przeciwko PD-1 zatwierdzony przez FDA w lipcu 2017 r. po pomyślnych wynikach badania fazy II CheckMate 142 dotyczącego leczenia drugiej linii pacjentów z dMMR-MSI-H mCRC, u których wystąpił postęp choroby podczas leczenia oksaliplatyną, fluoropirymidyną i irynotekanem. W badaniu tym zgłoszono, że po 12 miesiącach obserwacji obiektywny wskaźnik odpowiedzi wystąpił u 31% pacjentów i 69% osób z grupy kontrolnej. 12-miesięczne przeżycie bez progresji (PFS) wyniosło 50%, a całkowite przeżycie (OS) 73%. Najczęstszymi działaniami niepożądanymi związanymi z terapią były świąd, wysypka, biegunka i zmęczenie. Mutacje BRAF, KRAS i ekspresja PD-L1 nie miały wpływu na odpowiedź na przepisaną terapię celowaną ( 85 ).
Drugim ICI zatwierdzonym przez FDA w maju 2017 r. był Pembrolizumab, substancja przeciwko PD-1, której skuteczność została udowodniona w pierwszej fazie badania Keynote 016. Początkowo wykazano, że pacjenci z mCRC dMMR-MSI-H doświadczyli 40% wskaźnika odpowiedzi (RR), podczas gdy pacjenci z pMMR-MSI-L – 0% RR. Później udokumentowano, że 2-letni PFS wynosił 53% w pierwszej grupie. Ciężkie działania niepożądane wystąpiły tylko u 14% pacjentów, w tym trombocytopenia, leukopenia i zapalenie trzustki. Ta lecznicza opcja monoterapii została przepisana pacjentom z dMMR-MSI-H mCRC, których stan pogorszył się w trakcie lub po terapii oksaliplatyną, fluoropirymidyną i irynotekanem.
Następnym podejściem immunoterapeutycznym zatwierdzonym przez FDA w przypadku opornego CRC, który rozwinął się podczas terapii oksaliplatyną, fluoropirymidyną i irynotekanem, było połączenie niwolumabu z ipilimumabem (środek anty-CTLA-4). Zatwierdzenie zostało przyznane w lipcu 2018 r., po raporcie wyników badania fazy II CheckMate 142. Podczas obserwacji po 13,4 miesiącach obiektywny wskaźnik odpowiedzi wyniósł 54,7%, przy częściowej odpowiedzi −51,3%, całkowitej odpowiedzi −3,4%, a wskaźnik kontroli choroby przez 3 miesiące lub dłużej −80%. PFS po 12 miesiącach wyniósł 71%, OS −85%. Trzynaście procent pacjentów było zmuszonych przerwać leczenie z powodu działań niepożądanych związanych z lekiem. Ta kombinacja wykazała lepszą skuteczność niż monoterapia anty-PD-1. Jednakże działania niepożądane stopnia 3–4 były bardziej widoczne w przypadku terapii skojarzonej w porównaniu z leczeniem jednym lekiem i wynosiły odpowiednio 32–20% ( 86 ).
W przypadku pacjentów z pMMR-MSI-L należy przeprowadzić więcej badań nad schematami immunoterapeutycznymi. Istnieje potrzeba znalezienia leków, które będą ukierunkowane na odpowiedź immunologiczną, a także będą promować naciek komórek T. Ze względu na niskie obciążenie mutacjami i neoantygenami trudno jest osiągnąć te cele. Obecne schematy obejmują radioterapię, chemioterapię i substancje antyangiogenne w celu zwiększenia aktywacji immunologicznej, zabijania komórek nowotworowych i podwyższenia antygenów nowotworowych. Później leczenie można łączyć z ICI i innymi lekami biologicznymi. Obecnie trwają pewne badania kliniczne, które oceniają skutki chemioterapii z zastosowaniem terapii anty-PD-1, anty-PD-L1 i zewnętrznej radioterapii wiązką lub ablacji częstotliwości radiowej ( 87 ). Trwające badanie NCT01633970 fazy Ib oceniało skuteczność atezolizumabu (anty-PD-L1) i bevacizumabu plus FOLFOX; wykazało ono OS na poziomie 7% i stabilizację choroby u 64% pacjentów. Innym dobrze przebadanym podejściem jest połączenie inhibitorów kinazy białkowej aktywowanej mitogenem (MEK), takich jak Cobimetinib i Atezolizumab. Inhibitory MEK mogą dodatkowo uwrażliwić MSS mCRC na terapię celowaną. Badanie kliniczne fazy Ib ( NCT01988896 ) oceniło to połączenie u pacjentów z opornym na leczenie mutantem KRAS CRC i pMMR-MSI-L CRC i wykazało RR wynoszący 17%, gdzie pięciu pacjentów z 23 miało stabilną chorobę, a u czterech pacjentów rozwinęła się PR. Nie odnotowano żadnych zaawansowanych działań niepożądanych związanych z terapią. Później włączono 84 pacjentów, a wyniki zaktualizowano. RR wyniosło 8%, wskaźnik kontroli choroby -−31%. 6-miesięczne PFS i 12-miesięczne OS wyniosły odpowiednio 27 i 51%. To podejście jest bardzo obiecujące, ponieważ pokazuje, że inhibitory MEK mogą zwiększyć odpowiedź na immunoterapię u pacjentów z MSS mCRC. Podczas trwającego badania NCT03406871 , w którym połączono niwolumab i regofarenib (inhibitor wielokinazowy), przedstawiono obiecujące wyniki ; u 18 z 19 pacjentów wystąpiła obiektywna odpowiedź guza (siedem z nich to MSS CRC, 11 — rak żołądka MSS i 1 — MSI-H CRC). Nadal konieczne są bardziej spersonalizowane podejścia do leczenia pMMR-MSI-L ( 84 ).
Znaczenie ECS dla CRC
ECS aktywnie reguluje homeostazę jelit. Wszystkie składniki ECS są silnie wyrażone w tkance jelitowej, co oznacza, że ten system bezpośrednio wpływa na prawidłowe funkcjonowanie układu żołądkowo-jelitowego. Receptory CB1 i CB2 są wyrażone w zdrowym nabłonku okrężnicy, podśluzówkowym splocie mięśniowo-jelitowym i mięśniach gładkich, komórkach plazmatycznych w blaszce właściwej; receptor CB2 jest również obecny na makrofagach jelitowych ( 88 , 89 ). Receptor TRPV1 jest wyrażany na włóknach nerwowych okrężnicy ( 90 ). Receptor GPR55 jest obecny w błonie śluzowej i warstwie mięśniowej okrężnicy ( 91 ). Endokannabinoidy, 2-AG i AEA są również obecne w zdrowej tkance okrężnicy ( 92 ). Główne enzymy degradujące endokannabinoidy, enzymy FAAH i MAGL są rozmieszczone na gruczołach nabłonka okrężnicy, blaszce właściwej i splocie mięśniowo-jelitowym. Enzymy biosyntezy NAPE-PLD i DAGL występują na mięśniach gładkich jelita grubego, blaszce właściwej i gruczołach nabłonkowych; DAGL występuje również na splocie mięśniówkowym jelita ( 89 ).
Aby zrozumieć rolę ECS w jelitach, ważne jest rozróżnienie skutków zwiększonego i zmniejszonego tonu kannabinoidowego w układzie żołądkowo-jelitowym. Ogólnie rzecz biorąc, antagoniści receptora CB1 zmniejszają ton kannabinoidowy w jelitach i prowadzą do wymiotów, biegunki, zwiększonego opróżniania żołądka i pasażu żołądkowo-jelitowego. Natomiast agoniści receptora CB1 i CB2, a także inhibitory MAGL i blokery FAAH prowadzą do zwiększenia tonu kannabinoidowego jelit poprzez zmniejszenie wymiotów, wydzielania kwasu żołądkowego i opróżniania żołądka, a także zmniejszenie nadruchliwości, biegunki i bólu trzewnego ( 93 ). Wyciszenie receptora CB1 przez selektywnego antagonistę receptora CB1 AM251 u myszy ApcMin/+ doprowadziło do zwiększenia liczby polipów jelitowych, podczas gdy aktywacja receptora CB1 spowodowała śmierć komórek nowotworowych. Natomiast wyciszenie receptora CB2 nie wykazało żadnego wpływu na wzrost polipów ( 94 ).
Składniki ECS są znacząco rozregulowane w CRC. Endokannabinoidy (2-AG i AEA) były 3-krotnie wyższe w gruczolakach i 2-krotnie wyższe w CRC w porównaniu do prawidłowej błony śluzowej jelita grubego ( 92 ). Ekspresja receptora CB1 jest zmniejszona w CRC ( 95 ). Ekspresja receptora CB2 jest zwiększona w CRC i jest uważana za zły czynnik prognostyczny w tym typie raka ( 96 ). Poziomy FAAH i MAGL były również zwiększone u pacjentów z CRC ( 97 ). ECS jest bardzo ważnym czynnikiem patogenezy CRC, co sugeruje potencjalny wpływ kannabinoidów na tę chorobę.
Roślina lecznicza , która ostatnio zyskała wiele uwagi w dziedzinie leczenia raka, to Cannabis sativa . Wiele eksperymentów in vitro i in vivo wykazało, że kannabinoidy i ekstrakty z konopi hamują proliferację, stymulują apoptozę i autofagię, tłumią angiogenezę i przerzuty ( 98–100 ). Głównymi aktywnymi kannabinoidami odpowiedzialnymi za te efekty są kannabigerol (CBG), kannabidiol (CBD) i tetrahydrokannabinol (THC). Wykazano, że CBG aktywował apoptozę, stymulował produkcję ROS, zwiększał mRNA CHOP i hamował wzrost komórek w komórkach CRC (Caco-2, HCT-116) ( 101 ). Stwierdzono, że hamujący wpływ CBG na żywotność komórek raka jelita grubego zależał od czasu. W wyciszonych komórkach TRPM8 hamujący wpływ CBG na wzrost komórek był wyraźnie tłumiony w porównaniu z komórkami niewyciszonymi. Indukcję apoptozy wykazano poprzez wzrost aktywności kaspaz 3 i 7, obecność fragmentów DNA, wzrost ekspresji CHOP. W tym samym artykule wykazano, że CBG (3 lub 10 mg/kg) hamowało wzrost guzów ksenoprzeszczepowych (HCT-116) w modelu myszy o 45,3% i chemicznie indukowało karcynogenezę jelita grubego w modelach za pomocą azoksymetanu (AOM), w którym CBG w stężeniu 5 mg/kg całkowicie hamowało powstawanie nieprawidłowych ognisk krypt (ACF), zmniejszało liczbę guzów o połowę i nie wpływało na powstawanie polipów ( 101 ).
Wykazano również, że CBD ma działanie antyproliferacyjne w modelach raka jelita grubego. W niektórych badaniach in vitro CBD chroniło DNA przed stresem oksydacyjnym, podnosiło poziom endokannabinoidów i hamowało proliferację komórek raka jelita grubego za pośrednictwem receptorów CB1, TRPV1, PPAR-γ ( 102 ). Selektywni antagoniści rimonabant i AM251 (antagonista CB1R), kapsazepina (antagonista TRPV1R), GW 9662 (antagonista receptora PPAR-γ) hamowali działanie antyproliferacyjne CBD. Chemoprewencja CBD została potwierdzona przy użyciu modeli in vivo raka jelita grubego wywołanego AOM. CBD (1 mg/kg) zmniejszyło ACF o 67%, liczbę guzów o 66% i polipów o 57%. Gdy stężenie wzrosło do 5 mg/kg, zapobiegało jedynie tworzeniu się polipów. Efekt ten był spowodowany aktywacją kaspazy-3 i zmniejszeniem fosforylowanej formy białka Akt ( 102 ). W innym badaniu wykazano proapoptotyczny efekt CBD w komórkach CRC (HCT-116, DLD-1) i zasugerowano, że jest on wynikiem aktywacji Noxa, zwiększenia produkcji ROS i indukcji stresu siateczki śródplazmatycznej. Gdy poziomy Noxa zostały stłumione przez siRNA, ekspresja markerów apoptozy uległa znacznemu zmniejszeniu. Podobnie, po zablokowaniu produkcji ROS, poziom Noxa został zmniejszony. CBD indukował apoptozę w sposób zależny od Noxa-ROS ( 103 ). Ponadto, podczas stosowania raka jelita grubego indukowanego linią komórkową CT26 u myszy, CBD w stężeniach 1 i 5 mg/kg wykazywało działanie przeciwangiogenne i przeciwprzerzutowe poprzez hamowanie VEGF, przy czym ta druga dawka była skuteczniejsza. U zwierząt otrzymujących CBD zaobserwowano istotny wzrost aktywności enzymów antyoksydacyjnych, w tym SOD, GPX, GR, TAC i spadek MDA ( 104 ).
Badano również wpływ pełnych ekstraktów botanicznych, takich jak botaniczna substancja lecznicza o wysokiej zawartości CBD (BDS), na raka jelita grubego. Takie ekstrakty są zazwyczaj przygotowywane z kwiatów konopi, które są bogate w CBD, lub izolat CBD jest dodawany (wzbogacany) do określonego stężenia. Postawiono hipotezę, że inne składniki ekstraktów z roślin konopi mogą działać synergicznie z CBD i mogą być przydatne z terapeutycznego punktu widzenia. Wykazano, że CBD BDS ma znaczące właściwości antyproliferacyjne na komórki rakowe (HCT-116, DLD-1), podczas gdy zdrowe komórki nabłonka jelita grubego nie zostały dotknięte. Nie odnotowano różnicy w sile i skuteczności między CBD BDS a CBD, gdy stosowano te same dawki (0,3–5 μM). Efekty CBD BDS były neutralizowane przez selektywnych antagonistów receptorów CB1 i CB2. CBD BDS miało wyraźniejsze powinowactwo do receptorów CB1 i CB2 niż czysty CBD. Badania in vivo wykazały, że zastosowanie chemicznie indukowanej karcynogenezy przez AOM, ekstraktu z C. sativa o wysokiej zawartości kannabidiolu, zahamowało ACF o 86%, polipy o 79% i tworzenie się guzów o 40%. W modelach ksenoprzeszczepu CBD BDS znacząco zmniejszyło objętość guza, ale nie zaobserwowano różnicy w rozwoju guzów po 1 tygodniu leczenia ( 105 ).
Wykazano, że THC indukuje apoptozę w komórkach raka jelita grubego poprzez aktywację receptorów CB1 i hamowanie PI3K-AKT, kaskady RAS-MAPK i aktywacji BAD. Komórki raka jelita grubego (SW480, HCT-15, HT29, Caco-2, HCT-116 i SW620) wystawione na działanie THC (2,5–12,5 μM) skutkowały dawkozależnym zmniejszeniem przeżywalności komórek. Natomiast mniejsze stężenia od 100 nM do 1 μM nie miały zauważalnego wpływu na proliferację i przeżywalność komórek raka jelita grubego. THC zwiększało poziomy kaspazy-3 i PARP (substratu kaspazy-3). THC powodowało defosforylację i aktywację BAD ( 106 ). Potencjał przeciwnowotworowy kannabinoidów w CRC podsumowano w Tabeli 4 .
Tabela 4.
Potencjał przeciwnowotworowy kanabinoidów w raku jelita grubego.
Fitokannabinoidy
Typ modelu
Działanie przeciwnowotworowe/mechanizm działania
Odniesienia
CBG
in vitro (Caco-2, HCT116) in vivo (samice myszy grasicy, ksenograft-HCT116; model raka jelita grubego wywołanego azoksymetanem)
Proapoptotyczne, antyproliferacyjne; pobudza produkcję ROS, zwiększa mRNA CHOP, zwiększa poziom aktywności kaspazy 3, 7; w guzach ksenoprzeszczepionych – zmniejsza wzrost guza, w guzach AOM – całkowicie hamuje ACF, zmniejsza liczbę guzów
Powolny rozwój i zatwierdzanie nowych leków przeciwnowotworowych na CRC wynika z braku odpowiednich modeli przedklinicznych. Modele 2D in vitro umożliwiają przeprowadzanie badań przesiewowych o wysokiej przepustowości i są proste w obsłudze, ale pozwalają jedynie na badanie interakcji komórka-komórka lub komórka-matryca, a nie całego TME; dlatego nie są fizjologicznie istotne i nie są klinicznie predykcyjne. Z drugiej strony, modele zwierzęce in vivo pozwalają na badanie interakcji całego organizmu z odpowiednim TME i heterogenicznością wewnątrzguzową, ale te modele nie nadają się do badań przesiewowych na dużą skalę, są bardzo czasochłonne i nie są „ludzkie”. Tak więc zarówno modele in vitro , jak i in vivo stanowią cenne narzędzie do badania karcynogenezy jelita grubego ( 107 ). Jednak ze względu na wspomniane różnice korelacja między tymi modelami nie jest zbyt silna ( 108 ). Z drugiej strony badania kliniczne są złotym standardem testowania i zatwierdzania każdego potencjalnego leku.
Ważne jest, aby wspomnieć o jednym badaniu klinicznym, które zbadało największą liczbę pacjentów onkologicznych otrzymujących medyczną marihuanę w latach 2015-2017 w Izraelu. Dwa tysiące dziewięćset siedemdziesięciu pacjentów cierpiących na raka piersi (20,7%), płuc (13,6%), trzustki (8,1%) i jelita grubego (7,9%) otrzymywało medyczną marihuanę jako leczenie paliatywne w celu złagodzenia objawów, takich jak ból, brak apetytu, złe samopoczucie, zaburzenia snu i nudności. W tym badaniu wykorzystano cztery rodzaje marihuany: szczepy sativa o wysokiej zawartości THC, bez CBD; szczepy indica o wysokiej zawartości THC bez CBD; szczep z równą ilością CBD i THC oraz szczepy bogate w CBD. Co ciekawe, większość pacjentów otrzymywała więcej niż jeden szczep. Dziewięćset dwóch (24,9%) pacjentów zmarło, a 682 (18,8%) pacjentów przerwało leczenie po 6 miesiącach obserwacji. Spośród pozostałych pacjentów 60,6% odpowiedziało na leczenie; U 95,9% pacjentów nastąpiła znacząca poprawa stanu, u 3,7% — brak zmian, u 0,3% — pogorszenie. Przed rozpoczęciem leczenia tylko 18,7% pacjentów stwierdziło dobrą jakość życia, natomiast po 6 miesiącach leczenia —69,5%. Spośród wszystkich objawów związanych z rakiem najbardziej poprawiły się nudności, wymioty, depresja, migrena i zaburzenia snu. Najczęstszymi skutkami ubocznymi leczenia konopiami po 6 miesiącach obserwacji były zawroty głowy, suchość w jamie ustnej i zwiększony apetyt. Psychoaktywne działania niepożądane zauważyło tylko 2,8% pacjentów. Co ciekawe, spośród 344 pacjentów przyjmujących opioidy, 36% z nich przerwało ich przyjmowanie. Stwierdzono, że medyczna marihuana jest dobrze tolerowaną i bezpieczną opcją terapii paliatywnej dla pacjentów onkologicznych ( 109 ).
Rola konopi w wrodzonej i adaptacyjnej odpowiedzi immunologicznej
Będąc środkami immunomodulującymi, ekstrakty z konopi i pojedyncze kannabinoidy mogą wpływać zarówno na wrodzoną, jak i adaptacyjną odpowiedź immunologiczną. Ogólnie rzecz biorąc, kannabinoidy są uważane za związki immunosupresyjne. Wpływają na wrodzoną odpowiedź immunologiczną poprzez hamowanie aktywności komórek NK, komórek dendrytycznych, migrację neutrofili i makrofagów z ich procesami prezentacji antygenu i fagocytozy ( 110 ) oraz poprzez wyzwalanie indukcji MDSC ( 111 , 112 ). Zapalenie jest głównym mechanizmem wrodzonej odpowiedzi immunologicznej. Ogólnie rzecz biorąc, kannabinoidy, takie jak THC i CBD, powodują zmniejszenie ekspresji cytokin prozapalnych i zwiększenie ekspresji cytokin przeciwzapalnych. Dzięki temu aktywnie hamują proces zapalny ( 57 ). Jednak niektóre badania wykazują, że związki te mają różny wpływ na stan zapalny, albo go wzmacniając, albo tłumiąc. Na przykład CBD może aktywować odpowiedź immunologiczną poprzez podniesienie ekspresji mRNA TNF-α, IL-6, jak wykazano u myszy w odpowiedzi na zapalenie płuc wywołane LPS ( 113 ). Natomiast CBD hamowało IL-6 i IL-8 w mysim modelu raka jelita grubego in vivo opartym na linii komórkowej CT26 ( 104 ). Te sprzeczne wyniki mogą być specyficzne dla tkanki i dawki.
Kanabinoidy mogą wpływać na adaptacyjne odpowiedzi immunologiczne poprzez oddziaływanie na odporność humoralną i komórkową. Odporność komórek T może być pod wpływem kanabinoidów na różne sposoby: mogą one wpływać na proliferację i liczbę komórek T poprzez polaryzację odpowiedzi cytokinowej na Th1 lub Th2 ( 114 ). Wykazano, że kanabinoidy hamują proliferację komórek T, powodują ich apoptozę i wspierają polaryzację Th2 ( 115 , 116 ). Niektóre wstępne badania in vitro i in vivo THC wykazały immunosupresyjny wpływ na komórki T i komórki B przy stosowaniu wysokich stężeń, podczas gdy efekty immunostymulujące obserwowano przy niskich stężeniach ( 110 ). Badania eksperymentalne przeprowadzone in vivo na makakach zakażonych wirusem SIV, które otrzymywały THC przez okres 17 miesięcy, wykazały wzrost liczby komórek T, zmniejszenie ładunku wirusowego i wzrost ekspresji cytokin Th2 ( 117 ). Inne badanie przeprowadzone na pacjentach z HIV wykazało wyższe stężenie komórek CD4+ i CD8+ u pacjentów z THC-dodatnim wynikiem w porównaniu z pacjentami z THC-ujemnym wynikiem ( 118 ). Jeśli chodzi o rolę CBD, wykazano również, że może ono działać jako immunosupresant Th2 in vitro i in vivo , polaryzując odpowiedź cytokinową na Th2 i działając jako immunostymulant na Th1 ( 119 ). Jeśli chodzi o odporność humoralną, niektóre raporty z badań na ludziach wykazały zmniejszoną liczbę limfocytów B i zmniejszoną ilość IgM i IgG po spożyciu kannabinoidów w postaci bhang ( 120 ).
Przyszłe perspektywy wzmocnienia immunoterapii za pomocą kannabinoidów i ekstraktów z Cannabis Sativa
Immunomodulacyjne działanie konopi jest dobrze udokumentowane. Obecnie istnieje wiele znanych odmian konopi, a każda z nich ma unikalny skład różnych związków. Wiele badań wykazało wpływ pojedynczych kannabinoidów, takich jak THC i CBD, na stany zapalne i wzrost komórek rakowych ( 98 ). Inne składniki rośliny (takie jak mniejsze kannabinoidy, terpeny, terpenoidy, flawonoidy i inne) mogą działać synergicznie z kannabinoidami i mogą być przydatne z terapeutycznego punktu widzenia. Modulujący wpływ tych związków jest znany jako „efekt otoczenia”; taka modulacja jest zazwyczaj dodatnia, co oznacza, że efekt leczniczy całego ekstraktu z rośliny jest bardziej znaczący niż efekt izolowanych związków ( 121 ). Podobnie jak w przypadku każdego innego leku, efekty w znacznym stopniu zależą od stężenia. W przyszłości, dzięki dalszym badaniom, możemy uzyskać więcej informacji na temat potencjalnego immunostymulującego wpływu pojedynczych kannabinoidów lub ekstraktów z konopi. Wiedza ta może pomóc personelowi medycznemu w integracji ekstraktów z konopi z ukierunkowaną terapią nowotworową, potencjalnie jako terapia wspomagająca. Należy zidentyfikować specjalne ekstrakty o silnym działaniu przeciwnowotworowym, które nie są cytotoksyczne dla normalnych komórek i mogą uwrażliwiać komórki nowotworowe na dalsze leczenie bez zmniejszania odpowiedzi immunologicznej. Następnie te ekstrakty można połączyć z immunoterapią, a takie połączenie może mieć działanie synergistyczne. Wyniki retrospektywnej analizy przeprowadzonej u pacjentów z czerniakiem, rakiem nerki i niedrobnokomórkowym rakiem płuc, gdy konopie były stosowane w połączeniu z lekiem immunoterapeutycznym Nivolumab, wykazały zmniejszenie RR, ale nie zmiany w PFS i OS. Potrzebne są dalsze badania w celu zbadania możliwych interakcji między kannabinoidami a lekami immunoterapeutycznymi ( 122 ).
Należy przeprowadzić dogłębną eksplorację badań nad konopiami i powiązanymi lekami. Obecnie dysponujemy ograniczonymi danymi na temat interakcji konopi z innymi lekami, zwłaszcza w przypadku terapii ukierunkowanej. Ponieważ inhibitory punktów kontrolnych układu odpornościowego są rodzajem najbardziej skutecznej i efektywnej immunoterapii dla pacjentów z rakiem jelita grubego. Dlatego też należy przeprowadzić badania nad możliwością wzmocnienia immunoterapii za pomocą ekstraktów z konopi ( Rysunki 2 , 3 ).
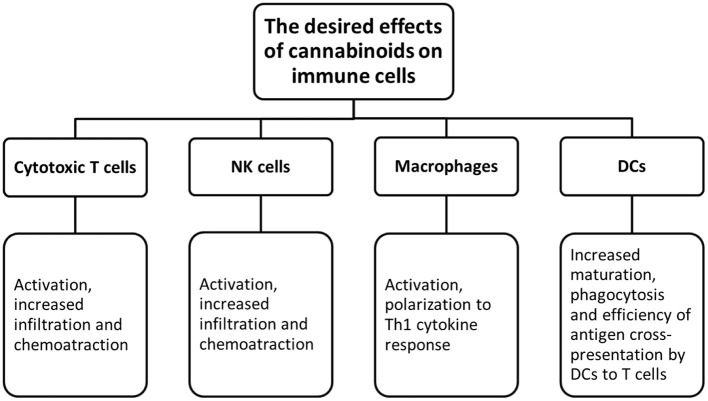
Rysunek 2.
Potencjał kannabinoidów w immunoterapii nowotworów. Górny panel pokazuje, jak kannabinoidy mogą zwiększyć immunogenność guza. Uwalnianie antygenów nowotworowych może być zwiększone ze względu na bezpośrednią cytotoksyczność kannabinoidów w komórkach nowotworowych. Następnie następuje prezentacja wzmocnionych antygenów nowotworowych, po której następuje wzrost odpowiedzi immunologicznej zależnej od limfocytów T i liza komórek nowotworowych przez limfocyty T. Dolny panel pokazuje, jak kannabinoidy mogą odwrócić immunosupresję guza. Makrofagi można przeprogramować do fenotypu przeciwnowotworowego za pomocą kannabinoidów. Immunostymulujące makrofagi M1 wydzielają cytokiny przeciwnowotworowe i skutecznie fagocytują komórki nowotworowe.
Rysunek 3.
Pożądane efekty kannabinoidów na komórki odpornościowe.
Pierwsze podejście mogłoby się skupić na znalezieniu ekstraktów, które mogą zwiększyć immunogenność guza. Z tego powodu komórki nowotworowe będą bardziej podatne na rozpoznanie przez układ odpornościowy. W takim przypadku badania in vivo byłyby korzystne, aby zbadać konopie indyjskie jako terapię neoadjuwantową przed rozpoczęciem stosowania leków biologicznych. Powinny być one następnie poddane badaniom sprawdzającym, czy występuje wzmocnienie adaptacyjnych odpowiedzi immunologicznych pośredniczonych przez limfocyty T. Poprzez kierowanie cytotoksyczności do komórek rakowych, ekstrakty z konopi indyjskich mogą zwiększyć uwalnianie antygenów nowotworowych, a następnie wzmocnić prezentację antygenu. Należy również ocenić zmiany w mikrośrodowisku guza, takie jak naciek MDSC i Treg. Drugie podejście mogłoby się skupić na możliwości odwrócenia immunosupresji wywołanej przez guz za pomocą ekstraktów. Na przykład można to zrobić, przeprogramowując makrofagi na fenotyp przeciwnowotworowy. Będąc wysoce plastycznymi komórkami, makrofagi mogą łatwo przejść z typu pro-nowotworowego na typ przeciwnowotworowy. Niektóre badania in vivo wykazały, że wpływanie na szlak PI3kγ w makrofagach może dalej prowadzić do ich polaryzacji do typu immunostymulującego poprzez zwiększenie cytotoksyczności komórek CD8+ T i poprawę odpowiedzi na ICIs ( 123 ). Znajdując ekstrakty, które mogą wpływać na ten konkretny szlak, można połączyć je z immunoterapią, aby wykazać działanie synergistyczne. Ponadto ocenę polaryzacji makrofagów można przeprowadzić poprzez badanie cytokin. Ekstrakty z konopi, które polaryzują makrofagi do typu M1 i nie posiadają właściwości przeciwzapalnych, można dalej łączyć z immunoterapią.
Wniosek
W literaturze silnie sugerowano, że kannabinoidy i ekstrakty z konopi mogą być stosowane w leczeniu raka jelita grubego. Dowody wskazują, że kannabinoidy mają duży potencjał przekształcenia się w obiecujące leki. Oczywiste jest, że związki te mogą oddziaływać na kluczowe szlaki sygnałowe rozwoju raka. Ponadto należy przeprowadzić więcej przedklinicznych i klinicznych ocen kannabinoidów jako środków przeciw CRC i immunomodulujących. Dodatkowe badania pomogłyby nam znaleźć nowe możliwości zapobiegawcze i terapeutyczne dla pacjentów, u których istnieje ryzyko rozwoju CRC lub którzy obecnie się z nim zmagają. Wprowadzając te silne związki do obecnych protokołów leczenia, można osiągnąć redukcję dawki innych leków, które są wysoce toksyczne, zmniejszając w ten sposób niepożądane skutki uboczne; podobnie kannabinoidy mogą prawdopodobnie uwrażliwić komórki złośliwe na dalszą ukierunkowaną terapię.
Ponadto dalsze badania nowotworów, zwłaszcza ich sekwencjonowanie, dostarczą więcej informacji na temat ich specyficznych cech. Wiedza o tym, w jaki sposób ścieżki genetyczne oddziałują z pewnymi kannabinoidami, może pomóc lekarzom przepisywać je zgodnie z genetycznym składem nowotworu. Z tego powodu personel medyczny będzie przepisywał terapie oparte na kannabinoidach konkretnemu pacjentowi z konkretną chorobą nowotworową. Leczenie stanie się zorientowane na pacjenta i specyficzne dla wskazań. W rezultacie rokowanie w przypadku nowotworu i jego wskaźnik przeżycia mogą się znacznie poprawić.
W świetle szybko zmieniającego się krajobrazu dotyczącego legalizacji marihuany do celów medycznych i rekreacyjnych, pacjenci mogą być bardziej skłonni pytać lekarzy o jej potencjalne negatywne i korzystne skutki dla zdrowia. Popularne przekonanie wydaje się być takie, że marihuana jest nieszkodliwą przyjemnością, do której dostęp nie powinien być regulowany ani uważany za nielegalny. Obecnie marihuana jest najczęściej używanym „nielegalnym” narkotykiem w Stanach Zjednoczonych, przy czym około 12% osób w wieku 12 lat i starszych zgłosiło używanie w ciągu ostatniego roku, a szczególnie wysokie wskaźniki używania występują wśród osób młodych. 1 Najczęstszą drogą podawania jest inhalacja. Zielonkawo-szare poszarpane liście i kwiaty rośliny Cannabis sativa są palone (wraz z łodygami i nasionami) w papierosach, cygarach, fajkach, fajkach wodnych lub „bluntach” (marihuana zwinięta w tytoniowy liść z cygara). Haszysz jest pokrewnym produktem wytwarzanym z żywicy kwiatów marihuany i jest zwykle palony (sam lub w mieszance z tytoniem), ale może być przyjmowany doustnie. Marihuanę można wykorzystać także do zaparzania herbaty, a wyciąg z jej oleju można dodawać do produktów spożywczych.
Regularne używanie marihuany w okresie dojrzewania jest szczególnie niepokojące, ponieważ używanie jej przez tę grupę wiekową wiąże się ze zwiększonym prawdopodobieństwem wystąpienia szkodliwych skutków 2 ( Tabela 1 ). Chociaż wiele badań wykazało szkodliwe skutki, inne nie, a kwestia, czy marihuana jest szkodliwa, pozostaje przedmiotem gorącej debaty. W niniejszym artykule dokonujemy przeglądu obecnego stanu wiedzy naukowej na temat niekorzystnych skutków zdrowotnych rekreacyjnego używania marihuany, skupiając się na tych obszarach, w których dowody są najsilniejsze.
Tabela 1.
Skutki uboczne krótkotrwałego oraz długotrwałego i intensywnego stosowania marihuany.
Skutki krótkotrwałego stosowania
Osłabiona pamięć krótkotrwała, utrudniająca uczenie się i zapamiętywanie informacji
Zaburzenia koordynacji ruchowej, utrudniające prowadzenie pojazdu i zwiększające ryzyko obrażeń
Zmiana osądu, zwiększająca ryzyko zachowań seksualnych, które ułatwiają przenoszenie chorób przenoszonych drogą płciową
W dużych dawkach paranoja i psychoza
Skutki długotrwałego lub intensywnego stosowania
Uzależnienie (u ok. 9% użytkowników ogółem, 17% osób, które zaczynają używać w okresie dojrzewania i 25–50% osób, które używają codziennie) *
Słabe wyniki w nauce, zwiększone prawdopodobieństwo porzucenia szkoły *
Osłabienie funkcji poznawczych, z niższym IQ wśród osób, które często korzystały z narkotyków w okresie dojrzewania *
Zmniejszone zadowolenie z życia i osiągnięcia (określone na podstawie miar subiektywnych i obiektywnych w porównaniu z ocenami w populacji ogólnej) *
Objawy przewlekłego zapalenia oskrzeli
Zwiększone ryzyko przewlekłych zaburzeń psychotycznych (w tym schizofrenii) u osób ze skłonnością do takich zaburzeń
DZIAŁANIA NIEPOŻĄDANE
RYZYKO UZALEŻNIENIA
Pomimo pewnych kontrowersyjnych dyskusji na temat uzależniającego działania marihuany, dowody jasno wskazują, że długotrwałe używanie marihuany może prowadzić do uzależnienia. Rzeczywiście, około 9% osób, które eksperymentują z marihuaną, uzależni się 3 (zgodnie z kryteriami uzależnienia zawartymi w Diagnostic and Statistical Manual of Mental Disorders , 4th edition [DSM-IV]). Liczba ta wzrasta do około 1 na 6 wśród osób, które zaczynają używać marihuany jako nastolatki i do 25-50% wśród osób, które palą marihuanę codziennie. 4 Zgodnie z Narodowym Badaniem Używania Narkotyków i Zdrowia z 2012 r. szacunkowo 2,7 miliona osób w wieku 12 lat i starszych spełniło kryteria DSM-IV dotyczące uzależnienia od marihuany, a 5,1 miliona osób spełniło kryteria dotyczące uzależnienia od jakiegokolwiek nielegalnego narkotyku 1 (8,6 miliona spełniło kryteria dotyczące uzależnienia od alkoholu 1 ). Istnieje również rozpoznanie prawdziwego zespołu odstawienia marihuany 5 (obejmującego objawy takie jak drażliwość, trudności ze snem, dysforia, głód i lęk), który utrudnia zaprzestanie i przyczynia się do nawrotu. Używanie marihuany przez nastolatków jest szczególnie kłopotliwe. Zwiększona podatność nastolatków na niekorzystne długoterminowe skutki używania marihuany jest prawdopodobnie związana z faktem, że mózg, w tym układ endokannabinoidowy, przechodzi aktywny rozwój w okresie dojrzewania. 6 Rzeczywiście, wczesne i regularne używanie marihuany przewiduje zwiększone ryzyko uzależnienia od marihuany, co z kolei przewiduje zwiększone ryzyko używania innych nielegalnych narkotyków. 7 W porównaniu z osobami, które zaczynają używać marihuany w wieku dorosłym, osoby, które zaczynają w okresie dojrzewania, mają około 2 do 4 razy większe prawdopodobieństwo wystąpienia objawów uzależnienia od marihuany w ciągu 2 lat od pierwszego użycia. 8
WPŁYW NA ROZWÓJ MÓZGU
Mózg pozostaje w stanie aktywnego, kierowanego doświadczeniem rozwoju od okresu prenatalnego przez dzieciństwo i okres dojrzewania do wieku około 21 lat. 9 W tych okresach rozwojowych jest on z natury bardziej podatny niż mózg dojrzały na niekorzystne, długoterminowe skutki czynników środowiskowych, takich jak ekspozycja na tetrahydrokanabinol, lub THC, główny składnik aktywny marihuany. Pogląd ten uzyskał znaczne poparcie w badaniach na zwierzętach, które wykazały na przykład, że prenatalna lub młodzieńcza ekspozycja na THC może ponownie skalibrować wrażliwość układu nagrody na inne narkotyki 10 i że prenatalna ekspozycja zakłóca dynamikę cytoszkieletu, która jest krytyczna dla ustanowienia połączeń aksonalnych między neuronami. 11
W porównaniu z osobami nienarażonymi na działanie marihuany, dorośli, którzy regularnie palili marihuanę w okresie dojrzewania, mają upośledzoną łączność nerwową (mniej włókien) w określonych obszarach mózgu. Należą do nich przedklinek, kluczowy węzeł zaangażowany w funkcje wymagające wysokiego stopnia integracji (np. czujność i samoświadomość), a także strzępki, obszar hipokampa, który jest ważny w uczeniu się i zapamiętywaniu. 12 Zmniejszona łączność funkcjonalna została również zgłoszona w sieciach przedczołowych odpowiedzialnych za funkcje wykonawcze (w tym kontrolę hamującą) i sieciach podkorowych, które przetwarzają nawyki i rutyny. 13 Ponadto badania obrazowe u osób używających marihuany wykazały zmniejszoną aktywność w obszarach przedczołowych i zmniejszone objętości w hipokampie. 14 Tak więc niektóre obszary mózgu mogą być bardziej podatne niż inne na długotrwałe skutki marihuany. Jedno z badań wykazało, że selektywne zmniejszenie ekspresji receptorów kannabinoidowych-1 (CB1) w kilku obszarach kory mózgowej u długotrwałych palaczy marihuany było powiązane z wieloletnim paleniem konopi i ustępowało po 4 tygodniach abstynencji. 15 Zmian w receptorach CB1 nie zaobserwowano w obszarach podkorowych.
Dane epidemiologiczne i przedkliniczne sugerują, że używanie marihuany w okresie dojrzewania może wpływać na wiele zachowań uzależniających w wieku dorosłym. U gryzoni narażonych na kannabinoidy w okresie dojrzewania występuje zmniejszona reaktywność neuronów dopaminowych, które modulują obszary nagrody w mózgu. 18 Narażenie gryzoni na działanie konopi w macicy zmienia regulację rozwojową układu dopaminowego mezolimbicznego u dotkniętego tym problemem potomstwa. 19 Jeśli zmniejszona reaktywność dopaminy w obszarach nagrody w mózgu następuje po wczesnym narażeniu na działanie marihuany, efekt ten może pomóc wyjaśnić zwiększoną podatność na nadużywanie narkotyków i uzależnienie od kilku narkotyków w późniejszym życiu, o czym donoszono w większości badań epidemiologicznych. 20 Ta teoria jest również zgodna z modelami zwierzęcymi, które pokazują, że THC może przygotowywać mózg do wzmocnionych reakcji na inne narkotyki. 21 Chociaż wyniki te potwierdzają tezę, że marihuana jest narkotykiem wprowadzającym, inne narkotyki, takie jak alkohol i nikotyna, również można zaliczyć do tej kategorii, ponieważ one również przygotowują mózg na wzmożoną reakcję na inne narkotyki. 22 Jednak alternatywne wyjaśnienie jest takie, że osoby, które są bardziej podatne na zachowania związane z przyjmowaniem narkotyków, po prostu częściej zaczynają od marihuany ze względu na jej dostępność, a ich późniejsze interakcje społeczne z innymi użytkownikami narkotyków zwiększają prawdopodobieństwo, że spróbują innych narkotyków.
ZWIĄZEK Z CHOROBĄ PSYCHICZNĄ
Regularne używanie marihuany wiąże się ze zwiększonym ryzykiem lęku i depresji, 23 ale związek przyczynowo-skutkowy nie został ustalony. Marihuana jest również powiązana z psychozami (w tym tymi związanymi ze schizofrenią), szczególnie u osób z istniejącą wcześniej podatnością genetyczną, 24 i zaostrza przebieg choroby u pacjentów ze schizofrenią. Intensywniejsze używanie marihuany, większa moc leku i narażenie w młodszym wieku mogą negatywnie wpłynąć na przebieg choroby (np. poprzez przyspieszenie czasu pierwszego epizodu psychotycznego o 2 do 6 lat). 25
Jednakże w tego typu badaniach trudno jest ustalić związek przyczynowo-skutkowy, ponieważ czynniki inne niż używanie marihuany mogą być bezpośrednio związane z ryzykiem choroby psychicznej. Ponadto inne czynniki mogą predysponować osobę zarówno do używania marihuany, jak i choroby psychicznej. Utrudnia to pewne przypisanie zwiększonego ryzyka choroby psychicznej używaniu marihuany.
WPŁYW NA WYNIKI SZKOLNE I OSIĄGNIĘCIA W CAŁYM ŻYCIU
W badaniu Monitoring the Future z 2013 r. wśród uczniów szkół średnich, 26 6,5% uczniów w klasie 12 zgłosiło codzienne lub prawie codzienne używanie marihuany, a liczba ta prawdopodobnie stanowi niedoszacowanie używania, ponieważ młodzi ludzie, którzy porzucili szkołę, mogą mieć szczególnie wysokie wskaźniki częstego używania marihuany. 27 Ponieważ używanie marihuany upośledza kluczowe funkcje poznawcze, zarówno podczas ostrego odurzenia, jak i przez kilka dni po użyciu, 28 wielu uczniów może funkcjonować na poziomie poznawczym, który jest poniżej ich naturalnych możliwości przez znaczne okresy czasu. Chociaż ostre efekty mogą ustąpić po usunięciu THC z mózgu, mimo to stwarza poważne ryzyko dla zdrowia, którego można się spodziewać przy długotrwałym lub intensywnym używaniu. Dowody sugerują, że takie używanie skutkuje mierzalnymi i długotrwałymi upośledzeniami poznawczymi, 16 szczególnie wśród tych, którzy zaczęli używać marihuany we wczesnej adolescencji. Co więcej, niezdolność do nauki w szkole, nawet przez krótkie lub sporadyczne okresy (wtórny skutek ostrego zatrucia), będzie miała wpływ na późniejszą zdolność do osiągania coraz trudniejszych celów edukacyjnych, co może również wyjaśniać związek między regularnym używaniem marihuany a słabymi ocenami.29
Związek między używaniem marihuany przez młodych ludzi a szkodami psychospołecznymi jest prawdopodobnie wieloaspektowy, co może wyjaśniać niespójności między badaniami. Na przykład, niektóre badania sugerują, że długoterminowe deficyty mogą być odwracalne i pozostać subtelne, a nie upośledzające, gdy dana osoba powstrzyma się od używania. 30 Inne badania pokazują, że długotrwałe, intensywne używanie marihuany powoduje upośledzenie pamięci i uwagi, które utrzymuje się i pogarsza wraz z kolejnymi latami regularnego używania31 i wraz z rozpoczęciem używania w okresie dojrzewania. 32 Jak zauważono powyżej, wczesne używanie marihuany wiąże się z pogorszeniem wyników w szkole i zwiększonym ryzykiem porzucenia szkoły, 27 , 29 chociaż doniesienia o wspólnych czynnikach środowiskowych, które wpływają na ryzyko używania marihuany w młodym wieku i porzucenia szkoły33 sugerują , że związek ten może być bardziej złożony. Intensywne używanie marihuany wiąże się z niższymi dochodami, większym zapotrzebowaniem na pomoc społeczno-ekonomiczną, bezrobociem, zachowaniami przestępczymi i niższym zadowoleniem z życia. 2 , 34
RYZYKO WYPADKÓW SAMOCHODOWYCH
Zarówno natychmiastowa, jak i długotrwała ekspozycja na marihuanę upośledzają zdolność prowadzenia pojazdów; marihuana jest nielegalnym narkotykiem najczęściej zgłaszanym w związku z upośledzoną jazdą i wypadkami, w tym wypadkami śmiertelnymi. 35 Istnieje związek między stężeniem THC we krwi a wynikami w kontrolowanych badaniach symulujących jazdę, 36 które są dobrym wskaźnikiem rzeczywistej zdolności prowadzenia pojazdów. Niedawne palenie marihuany i poziom THC we krwi wynoszący od 2 do 5 ng na mililitr są związane ze znacznym upośledzeniem zdolności prowadzenia pojazdów. 37 Zgodnie z metaanalizą, ogólne ryzyko udziału w wypadku wzrasta o współczynnik około 2, gdy osoba prowadzi pojazd wkrótce po użyciu marihuany. 37 W analizie winy za wypadek osoby z pozytywnym wynikiem testu na obecność THC (typowy minimalny poziom wykrywalności, 1 ng na mililitr), a w szczególności osoby z wyższym poziomem we krwi, były 3 do 7 razy bardziej narażone na spowodowanie wypadku samochodowego niż osoby, które nie używały narkotyków ani alkoholu przed prowadzeniem pojazdu. 38 Dla porównania, ogólne ryzyko wypadku samochodowego wzrasta prawie 5-krotnie w przypadku kierowców, u których stężenie alkoholu we krwi przekracza 0,08%, czyli dopuszczalny limit w większości krajów, a w przypadku osób poniżej 21. roku życia wzrasta 27-krotnie. 39 Nic dziwnego, że ryzyko związane ze stosowaniem alkoholu w połączeniu z marihuaną wydaje się być większe niż ryzyko związane ze stosowaniem każdego z tych narkotyków osobno. 37
RYZYKO RAKA I INNE SKUTKI DLA ZDROWIA
Wpływ długotrwałego palenia marihuany na ryzyko raka płuc jest niejasny. Na przykład używanie marihuany przez okres równoważny 30 lub więcej latom (przy czym 1 rok używania marihuany jest równy 1 papierosowi [skrętowi] marihuany wypalanemu dziennie przez 1 rok) wiązało się ze zwiększoną częstością występowania raka płuc i kilku nowotworów górnego odcinka przewodu pokarmowego; jednak związek ten zanikł po uwzględnieniu potencjalnych czynników zakłócających, takich jak palenie papierosów. 40 Chociaż nie można wykluczyć możliwości pozytywnego związku między paleniem marihuany a rakiem, 41 dowody sugerują, że ryzyko jest niższe w przypadku marihuany niż tytoniu. 40 Jednak palenie papierosów zawierających zarówno marihuanę, jak i produkty tytoniowe jest potencjalnym czynnikiem zakłócającym, którego rozpowszechnienie znacznie różni się w zależności od kraju.
Palenie marihuany wiąże się również ze stanem zapalnym dużych dróg oddechowych, zwiększonym oporem w drogach oddechowych i hiperinflacją płuc, co jest zgodne z faktem, że regularni palacze marihuany częściej zgłaszają objawy przewlekłego zapalenia oskrzeli niż osoby niepalące42 ; jednak długoterminowy wpływ niewielkich dawek marihuany nie wydaje się być znaczący.43 Kompetencja immunologiczna układu oddechowego u palaczy marihuany może być również upośledzona, o czym świadczy zwiększona częstość infekcji dróg oddechowych i zapalenia płuc.44 Używanie marihuany wiąże się również z chorobami naczyń, które zwiększają ryzyko zawału mięśnia sercowego, udaru i przemijających ataków niedokrwiennych podczas zatrucia marihuaną.45 Rzeczywiste mechanizmy leżące u podstaw wpływu marihuany na układ sercowo-naczyniowy i mózgowo-naczyniowy są złożone i nie do końca poznane. Jednakże bezpośrednie działanie kannabinoidów na różne receptory docelowe (np. receptory CB1 w tętniczych naczyniach krwionośnych) i pośrednie działanie na związki wazoaktywne46 może pomóc wyjaśnić szkodliwy wpływ marihuany na opór naczyniowy i mikrokrążenie wieńcowe.47
OGRANICZENIA DOWODÓW I LUKI W WIEDZY
Większość długoterminowych skutków używania marihuany, które są tutaj podsumowane, zaobserwowano u użytkowników intensywnie lub długoterminowo, ale liczne (często ukryte) czynniki zakłócające utrudniają nam ustalenie związku przyczynowo-skutkowego (w tym częste używanie marihuany w połączeniu z innymi narkotykami). Czynniki te utrudniają również ocenę prawdziwego wpływu wewnątrzmacicznej ekspozycji na marihuanę. Rzeczywiście, pomimo używania marihuany przez kobiety w ciąży, 48 i modeli zwierzęcych sugerujących, że ekspozycja na konopie w czasie ciąży może zmieniać normalne procesy i trajektorie rozwoju mózgu, 49 nasze zrozumienie długoterminowych skutków prenatalnej ekspozycji na marihuanę u ludzi jest bardzo słabe.
Zawartość THC, czyli moc marihuany, wykryta w skonfiskowanych próbkach, stale wzrastała z około 3% w latach 80. do 12% w 2012 r. 50 ( rys. 1A ). Ten wzrost zawartości THC budzi obawy, że konsekwencje używania marihuany mogą być teraz gorsze niż w przeszłości i może wyjaśniać znaczny wzrost liczby wizyt na oddziałach ratunkowych osób zgłaszających używanie marihuany 51 ( rys. 1B ) i wzrost liczby śmiertelnych wypadków samochodowych. 35 Ten wzrost mocy THC w czasie budzi również pytania o obecną trafność ustaleń starszych badań nad skutkami używania marihuany, zwłaszcza badań oceniających długoterminowe wyniki.
Rysunek 1. Wzrost stężenia tetrahydrokanabinolu (THC) w marihuanie w czasie oraz liczba wizyt na oddziale ratunkowym w związku z marihuaną, kokainą lub heroiną.
Wykres A przedstawia wzrastającą moc marihuany (tj. procentową zawartość THC) w próbkach skonfiskowanych przez Drug Enforcement Administration (DEA) w latach 1995–2012. Wykres B przedstawia szacunki liczby wizyt na oddziałach ratunkowych związanych z zażywaniem wybranych narkotyków (marihuany, kokainy i heroiny) pojedynczo lub w połączeniu z innymi narkotykami w latach 2004–2011. Spośród tych trzech narkotyków jedynie marihuana, stosowana w połączeniu z innymi narkotykami lub samodzielnie, wiązała się ze znacznym wzrostem liczby wizyt w tym okresie (wzrost o 62% w przypadku stosowania w połączeniu z innymi narkotykami i wzrost o 100% w przypadku stosowania samodzielnie, P<0,05 dla obu porównań).
Istnieje również potrzeba lepszego zrozumienia, w jaki sposób wykorzystać potencjalne korzyści medyczne rośliny marihuany bez narażania osób chorych na jej wewnętrzne ryzyko. Autorytatywny raport Instytutu Medycyny, Marihuany i Medycyny52 uznaje potencjalne korzyści palenia marihuany w pobudzaniu apetytu, szczególnie u pacjentów z zespołem nabytego niedoboru odporności (AIDS) i powiązanym zespołem wyniszczenia, a także w zwalczaniu nudności i wymiotów wywołanych chemioterapią , silnego bólu i niektórych form spastyczności. Raport wskazuje również, że istnieją pewne dowody na korzyści ze stosowania marihuany w celu zmniejszenia ciśnienia śródgałkowego w leczeniu jaskry. Niemniej jednak raport podkreśla znaczenie skupienia wysiłków badawczych na terapeutycznym potencjale syntetycznych lub farmaceutycznie czystych kannabinoidów.52 Niektórzy lekarze nadal przepisują marihuanę w celach leczniczych, pomimo ograniczonych dowodów na korzyści (patrz ramka ). Praktyka ta budzi szczególne obawy w odniesieniu do długotrwałego stosowania przez wrażliwe populacje . Na przykład istnieją pewne dowody sugerujące, że u pacjentów z objawami zakażenia wirusem niedoboru odporności ludzkiej (HIV) lub AIDS, używanie marihuany może w rzeczywistości zaostrzać deficyty poznawcze związane z HIV. 75 Podobnie, potrzeba więcej badań, aby zrozumieć potencjalne skutki używania marihuany na związane z wiekiem pogorszenie funkcji poznawczych w ogóle, a w szczególności na upośledzenie pamięci.
Stany kliniczne z objawami, które można złagodzić leczeniem marihuaną lub innymi kannabinoidami.*.
Jaskra
Wczesne dowody korzyści marihuany u pacjentów z jaskrą (chorobą związaną ze zwiększonym ciśnieniem w oku) mogą być zgodne z jej zdolnością do wywoływania przejściowego obniżenia ciśnienia śródgałkowego, 53 , 54 ale inne, standardowe metody leczenia są obecnie bardziej skuteczne. Wykazano, że THC, kannabinol i nabilon (syntetyczny kannabinoid podobny do THC), ale nie kannabidiol, obniżają ciśnienie śródgałkowe u królików. 55 , 56 Potrzebne są dalsze badania, aby ustalić, czy cząsteczki, które modulują układ endokannabinoidowy, mogą nie tylko obniżać ciśnienie śródgałkowe, ale także zapewniać korzyści neuroprotekcyjne u pacjentów z jaskrą. 57
Mdłości
Leczenie nudności i wymiotów związanych z chemioterapią było jednym z pierwszych zastosowań medycznych THC i innych kannabinoidów. 58 THC jest skutecznym środkiem przeciwwymiotnym u pacjentów poddawanych chemioterapii, 59 ale pacjenci często twierdzą, że marihuana jest skuteczniejsza w tłumieniu nudności. Inne, niezidentyfikowane związki w marihuanie mogą wzmacniać działanie THC (jak to się wydaje w przypadku THC i kannabidiolu, które działają poprzez różne mechanizmy przeciwwymiotne). 60 Paradoksalnie, zwiększone wymioty (hiperemesis) zgłaszano w przypadku powtarzającego się stosowania marihuany.
Anoreksja i zespół wyniszczenia związany z AIDS
Doniesienia wskazują, że palenie lub spożywanie konopi poprawia apetyt i prowadzi do zwiększenia masy ciała oraz poprawy nastroju i jakości życia u pacjentów z AIDS. 61 Jednakże nie ma długoterminowych lub rygorystycznych dowodów na trwały wpływ konopi na zachorowalność i śmiertelność związaną z AIDS, przy akceptowalnym profilu bezpieczeństwa, który uzasadniałby jej włączenie do obecnej praktyki klinicznej u pacjentów otrzymujących skuteczną terapię antyretrowirusową. 62 Dane z nielicznych badań, które badały potencjalną wartość terapeutyczną kannabinoidów dla tej populacji pacjentów, są niejednoznaczne. 62
Przewlekły ból
Marihuana była stosowana w celu łagodzenia bólu od stuleci. Badania wykazały, że kannabinoidy działające poprzez centralne receptory CB1, a prawdopodobnie także obwodowe receptory CB1 i CB2, 63 odgrywają ważną rolę w modelowaniu odpowiedzi nocyceptywnych w różnych modelach bólu. Wyniki te są zgodne z doniesieniami, że marihuana może być skuteczna w łagodzeniu bólu neuropatycznego, 64 , 65 nawet przy bardzo niskich poziomach THC (1,29%). 66 Zarówno marihuana, jak i dronabinol, farmaceutyczna formuła THC, zmniejszają ból, ale dronabinol może prowadzić do dłużej trwającego zmniejszenia wrażliwości na ból i niższych ocen efektów nagradzających. 67
Zapalenie
Kanabinoidy (np. THC i kannabidiol) mają znaczące działanie przeciwzapalne ze względu na zdolność do wywoływania apoptozy, hamowania proliferacji komórek i tłumienia produkcji cytokin. 68 Kanabidiol wzbudził szczególne zainteresowanie jako środek przeciwzapalny ze względu na brak efektów psychoaktywnych. 58 Modele zwierzęce wykazały, że kannabidiol jest obiecującym kandydatem do leczenia reumatoidalnego zapalenia stawów 58 i chorób zapalnych przewodu pokarmowego (np. wrzodziejącego zapalenia jelita grubego i choroby Leśniowskiego-Crohna). 69
Stwardnienie rozsiane
Nabiximols (Sativex, GW Pharmaceuticals), oromucosalny spray, który dostarcza mieszankę THC i kannabidiolu, wydaje się być skutecznym leczeniem bólu neuropatycznego, zaburzeń snu i spastyczności u pacjentów ze stwardnieniem rozsianym. Sativex jest dostępny w Wielkiej Brytanii, Kanadzie i kilku innych krajach 70 , 71 i jest obecnie poddawany przeglądowi w badaniach fazy 3 w Stanach Zjednoczonych w celu uzyskania zatwierdzenia od Food and Drug Administration.
Padaczka
W niedawnym małym badaniu rodziców, którzy stosują marihuanę o wysokiej zawartości kannabidiolu w leczeniu napadów padaczkowych u swoich dzieci, 72 11% (2 rodziny z 19, które spełniły kryteria włączenia) zgłosiło całkowity brak napadów, 42% (8 rodzin) zgłosiło zmniejszenie częstości napadów o ponad 80%, a 32% (6 rodzin) zgłosiło zmniejszenie częstości napadów o 25 do 60%. Chociaż takie raporty są obiecujące, nie ma wystarczających danych dotyczących bezpieczeństwa i skuteczności stosowania roślin konopi w leczeniu padaczki. 73 Jednak coraz więcej dowodów wskazuje na rolę kannabidiolu jako środka przeciwpadaczkowego w modelach zwierzęcych. 74
*AIDS oznacza zespół nabytego niedoboru odporności, receptor kannabinoidowy-1 CB1 i receptor kannabinoidowy-2 CB2, wirus niedoboru odporności ludzkiej HIV i tetrahydrokanabinol THC.
Potrzebne są badania nad sposobami, w jakie polityka rządowa dotycząca marihuany wpływa na wyniki w zakresie zdrowia publicznego. Nasza wiedza na temat wpływu polityki na siły rynkowe jest dość ograniczona (np. atrakcyjność nowych źródeł dochodów podatkowych z legalnej sprzedaży marihuany, wojny cenowe, reklamy skierowane do młodzieży i pojawienie się leków na bazie konopi zatwierdzonych przez Food and Drug Administration), podobnie jak nasza wiedza na temat powiązanych zmiennych postrzegania stosowania, rodzajów stosowania i wyników. Historycznie rzecz biorąc, istniała odwrotna korelacja między stosowaniem marihuany a postrzeganiem jej ryzyka wśród nastolatków ( rys. 2A ). Zakładając, że ta odwrotna zależność jest przyczynowa, czy większa pobłażliwość w kulturze i polityce społecznej doprowadziłaby do wzrostu liczby młodych ludzi, którzy są regularnie narażeni na działanie konopi? Wśród uczniów w klasie 12. zgłaszana częstość regularnego palenia marihuany stale rośnie w ostatnich latach i może wkrótce przeciąć linię trendu dla regularnego palenia tytoniu ( rys. 2B ). Potrzebujemy również informacji o skutkach biernego narażenia na dym z konopi i kannabinoidy. Bierne narażenie jest ważnym problemem zdrowia publicznego w kontekście palenia tytoniu, ale nie mamy jasnego zrozumienia skutków biernego narażenia na palenie marihuany. 76 Badania w stanach (np. Kolorado, Kalifornia i Waszyngton) i krajach (np. Urugwaj, Portugalia i Holandia), w których polityka społeczna i prawna ulega zmianie, mogą dostarczyć ważnych danych do kształtowania przyszłych polityk.
Rysunek 2. Używanie marihuany w odniesieniu do postrzeganego ryzyka i codziennego używania papierosów tytoniowych lub marihuany wśród uczniów amerykańskich klas 12. w latach 1975–2013.
Panel A pokazuje odwrotną korelację między postrzeganiem ryzyka związanego z używaniem marihuany a rzeczywistym używaniem. Postrzegane ryzyko odpowiada odsetkowi nastolatków, którzy zgłosili, że używanie marihuany jest niebezpieczne. Panel B pokazuje odsetek uczniów, którzy zgłosili codzienne używanie papierosów tytoniowych lub marihuany w ciągu ostatnich 30 dni. Dane dla obu wykresów pochodzą z Johnston et al. 26
WNIOSKI
Używanie marihuany wiąże się ze znacznymi skutkami ubocznymi, z których niektóre zostały określone z dużym prawdopodobieństwem ( Tabela 2 ). Marihuana, podobnie jak inne narkotyki, może powodować uzależnienie. Podczas odurzenia marihuana może zakłócać funkcje poznawcze (np. pamięć i postrzeganie czasu) i funkcje motoryczne (np. koordynację), a te efekty mogą mieć szkodliwe konsekwencje (np. wypadki samochodowe). Powtarzające się używanie marihuany w okresie dojrzewania może skutkować długotrwałymi zmianami w funkcjonowaniu mózgu, które mogą zagrozić osiągnięciom edukacyjnym, zawodowym i społecznym. Jednak skutki narkotyku (legalnego lub nielegalnego) na zdrowie jednostki są określane nie tylko przez jego właściwości farmakologiczne, ale także przez jego dostępność i akceptowalność społeczną. Pod tym względem legalne narkotyki (alkohol i tytoń) oferują trzeźwiącą perspektywę, odpowiadając za największe obciążenie chorobami związanymi z narkotykami 77 nie dlatego, że są bardziej niebezpieczne niż narkotyki nielegalne, ale dlatego, że ich status prawny pozwala na bardziej powszechną ekspozycję. W miarę jak polityka zmierza w kierunku legalizacji marihuany, rozsądne, a prawdopodobnie ostrożne, jest założenie, że wzrośnie jej użycie, a co za tym idzie, wzrośnie również liczba osób, u których wystąpią negatywne skutki zdrowotne.
Tabela 2.
Poziom zaufania do dowodów na negatywny wpływ marihuany na zdrowie i samopoczucie.
* Wskazany ogólny poziom pewności co do związku między używaniem marihuany a wymienionymi skutkami stanowi próbę oceny siły obecnych dowodów, zwłaszcza w odniesieniu do intensywnego lub długotrwałego używania oraz używania rozpoczynającego się w okresie dojrzewania.
Cel przeglądu: Otyłość brzuszna jest ściśle związana z cukrzycą typu 2 i nadaktywnością układu endokannabinoidowego. Niniejszy przegląd ma na celu ocenę roli układu endokannabinoidowego w dysregulacji glukozy oraz wpływu blokady receptora kannabinoidowego typu 1 na metabolizm glukozy zarówno w modelach zwierzęcych, jak i u ludzi z nadwagą/otyłością, zwłaszcza z cukrzycą typu 2.
Ostatnie odkrycia: Receptory kannabinoidowe typu 1 zostały zidentyfikowane nie tylko w mózgu, ale także w tkance tłuszczowej, jelitach, wątrobie, mięśniach szkieletowych, a nawet trzustce, wszystkich narządach odgrywających kluczową rolę w metabolizmie glukozy i cukrzycy typu 2. Rimonabant, pierwszy selektywny bloker receptora kannabinoidowego typu 1 stosowany klinicznie, wykazał skuteczność w redukcji masy ciała, obwodu talii, glikowanej hemoglobiny, triglicerydów, wskaźnika oporności na insulinę oraz zwiększeniu stężeń cholesterolu HDL i adiponektyny u pacjentów z cukrzycą typu 2, potwierdzając dane dotyczące pacjentów z nadwagą/otyłością, którzy nie mieli cukrzycy. Prawie połowa zmian metabolicznych, w tym redukcja glikowanej hemoglobiny, nie mogła być wyjaśniona tylko utratą wagi, zgodnie z bezpośrednimi efektami obwodowymi.
Podsumowanie: Blokada receptora kannabinoidowego typu 1 redukuje spożycie pokarmu i masę ciała, poprawiając regulację metaboliczną poza samą utratą wagi. Ze względu na pozytywny wpływ na metabolizm glukozy, rimonabant zasługuje na uwzględnienie w leczeniu pacjentów z nadwagą/otyłością i cukrzycą typu 2.
Optymalna homeostaza glukozy wymaga precyzyjnej adaptacji liczby komórek beta trzustki wydzielających insulinę w wyspach trzustki. Insulina pozytywnie reguluje proliferację komórek beta w sposób autokrynny poprzez szlak sygnalizacyjny receptora insuliny (IR). Wychodzi teraz na jaw, że aktywacja/antagonizm receptora kannabinoidowego typu 1 (CB1R) wpływa na działanie insuliny w tkankach wrażliwych na insulinę. Niemniej jednak nie zostało jednoznacznie ustalone, na których komórkach wyrażane są CB1R i jakie pełnią funkcje w wyspach trzustki. W ramach niniejszego badania przeprowadziliśmy badanie mające na celu sprawdzenie, czy endogenne kannabinoidy (ECs) wewnątrzwyspowe regulują proliferację komórek beta oraz czy wpływają na działanie insuliny.
METODY BADAŃ
Zmierzyliśmy produkcję ECs w izolowanych ludzkich i mysich wyspach oraz w linii komórkowej beta w odpowiedzi na glukozę i KCl. Oceniliśmy obecność w pełni funkcjonującego systemu EC w ludzkich i mysich wyspach, kilku liniach komórkowych beta oraz myszach z brakiem receptora CB1R (CB1R−/−). Zbadaliśmy, czy ECs wpływają na fizjologię komórek beta poprzez regulację działania insuliny i wykazaliśmy potencjał terapeutyczny manipulacji systemem EC u myszy diabetycznych (db/db).
WYNIKI
ECs są wytwarzane wewnątrz komórek beta, które również wyrażają CB1Rs, działające w pełni, gdy są aktywowane przez ligandy. Blokada genetyczna i farmakologiczna CB1R prowadzi do zwiększenia sygnalizacji IR przez szlak białka podścieliska insuliny 2-AKT w komórkach beta, co skutkuje zwiększeniem proliferacji i masy komórkowej beta. Antagonizm CB1R u myszy db/db prowadzi do obniżenia stężenia glukozy we krwi, zwiększenia proliferacji i masy komórkowej beta, oraz zwiększonej sygnalizacji IR w komórkach beta. Ponadto aktywacja CB1R utrudnia autodfosforylację IR indukowaną insuliną w komórkach beta w sposób zależny od podjednostki Gαi.
WNIOSKI
Te wyniki stanowią bezpośrednie dowody na funkcjonalne oddziaływanie między CB1R a sygnalizacją IR zaangażowaną w regulację proliferacji komórek beta i stanowić będą podstawę do opracowania nowych interwencji terapeutycznych w celu poprawy funkcji i proliferacji komórek beta w cukrzycy.
Endokannabinoidy i receptory kannabinoidowe CB1 są znane z ogólnej roli w homeostazie energetycznej. Jednak badania kliniczne z pierwszą generacją blokerów CB1, obecnie wycofane z powodu skutków ubocznych psychiatrycznych, początkowo miały na celu zmniejszenie spożycia jedzenia i masy ciała, a nie czynników ryzyka metabolicznego związanego z otyłością. W tym przeglądzie omawiamy, w jaki sposób, oprócz promowania spożycia energii, endokannabinoidy kontrolują metabolizm lipidów i glukozy w kilku narządach obwodowych, szczególnie w wątrobie i tkance tłuszczowej. Pojawiają się także bezpośrednie działania w mięśniach szkieletowych i trzustce. Ta wiedza może pomóc w projektowaniu przyszłych terapii zespołu metabolicznego.
Cel: Nowe pochodne kwasu tetrahydrokannabinolowego (THCA) o nazwie ALAM027 i ALAM108 zostały zaproponowane do leczenia nowotworu trzustki.
Metody: Wpływ nowych kannabinoidów ALAM027 i ALAM108 został zbadany in vitro na liniach komórkowych PANC-1 i AsPC-1 za pomocą testu CellTiter Glo. Do badania in vivo aktywności przeciwnowotworowej tych związków na komórkach PANC-1 wykorzystano model przeszczepu nowotworu trzustki.
Wyniki: Badanie in vitro nowych kannabinoidów wykazało większą aktywność ALAM108 niż ALAM027 zarówno w przypadku komórek PANC-1, jak i AsPC-1. Badanie in vivo nowych kannabinoidów na komórkach PANC-1 wykazało, że ich doustne podawanie było skuteczne w zmniejszaniu objętości i masy guza, nie powodując przy tym żadnych dolegliwości ani utraty wagi u myszy.
Wnioski: Kannabinoidy ALAM108 i ALAM027 hamowały wzrost guza 1,6–2 razy u myszy z ludzkimi komórkami PANC-1.
Słowa kluczowe: AsPC-1, kannabinoidy, in vitro, in vivo, PANC-1, THCA.
Rak trzustki to jedna z najniebezpieczniejszych form nowotworów ze względu na jego agresywny wzrost, wczesne przerzuty i słabą odpowiedź na wszelkie znane metody terapeutyczne. Stanowi obecnie czwartą najczęstszą przyczynę zgonów z powodu raka, a przewiduje się, że do 2030 roku stanie się drugą najczęstszą przyczyną zgonów z powodu nowotworów, zaraz po raku płuc, co wymaga pilnych działań w zakresie nowych strategii terapeutycznych, które mogą poprawić wyniki leczenia tej choroby.
Ostatnio rosnące zainteresowanie przeciwnowotworową aktywnością kannabinoidów doprowadziło do licznych badań obejmujących coraz więcej rodzajów nowotworów. Naturalne i syntetyczne kannabinoidy wykazały zdolność wpływania na proliferację, migrację i apoptozę komórek nowotworowych poprzez bezpośrednie i pośrednie aktywowanie receptorów kannabinoidowych CB1 i CB2. W przypadku raka trzustki wykazano, że ilość ekspresji receptorów CB1 i CB2 w komórkach nowotworowych jest znacznie wyższa niż w komórkach normalnych, co otwiera drogę do wykorzystania przeciwnowotworowych zdolności kannabinoidów do eliminacji komórek nowotworowych bez wpływu na normalne tkanki trzustki.
Dotychczas aktywność przeciwnowotworową kannabinoidów wobec guza trzustki była badana tylko w kilku badaniach przedklinicznych, które wykazały obiecującą aktywność przeciwnowotworową tetrahydrokannabinolu (THC) oraz niektórych kannabinoidów syntetycznych, takich jak WIN 55,212-2. Jednak patrząc w przyszłość i przechodząc do etapów klinicznych, THC jest mało prawdopodobne, aby było stosowane w leczeniu guzów ze względu na jego wyraźny efekt psychoaktywny. Jednocześnie badanie kannabinoidów o podobnej strukturze, ale pozbawionych właściwości psychoaktywnych, jest niewątpliwie interesujące. Jednym z obiecujących kierunków może być modyfikacja naturalnego kwasu tetrahydrokannabinolowego (THCA) poprzez syntezę jego pochodnych w grupie karboksylowej. Chociaż sam THCA wykazuje niewielką aktywność przeciwnowotworową wobec raka trzustki, modyfikacja fragmentu karboksylowego jego cząsteczki wykazała znaczny wzrost in vitro hamowania wzrostu komórek. W związku z tym dalsze modyfikacje grupy karboksylowej THCA mogą prowadzić do związków o bardziej specyficznym działaniu na komórki guza trzustki. Celem tej pracy jest zbadanie nowych pochodnych THCA, ALAM027 i ALAM108, na komórkach nowotworowych trzustki PANC-1 i AsPC-1.
Endokannabinoidy przez aktywację swoistych receptorów wpływają na regulację homeostazy energetycznej ustroju. Kontrolują m.in. pobór pokarmu, wydzielanie insuliny, metabolizm lipidów i glukozy, magazynowanie tłuszczów. Głównym substratem energetycznym mięśnia sercowego są długołańcuchowe kwasy tłuszczowe. Jednak serce ma ogromną elastyczność metaboliczną przejawiającą się zdolnością do utylizowania nie tylko kwasów tłuszczowych, ale także glukozy, mleczanów i ciał ketonowych. Endokannabinoidy mogą bezpośrednio oddziaływać na kardiomiocyty poprzez obecne na ich powierzchni receptory CB1 i CB2. Wydaje się, że bezpośrednia aktywacja receptorów CB1 sprzyja wzmożonej lipogenezie, stłuszczeniu osierdzia i zaburzeniom czynności bioelektrycznej serca. Natomiast pobudzone receptory CB2 wykazują właściwości kardioprotekcyjne, przyczyniając się do zachowania odpowiedniego stężenia ATP w kardiomiocytach. Ponadto, działanie endokannabinoidów zarówno na poziomie ośrodkowego układu nerwowego, jak i tkanek obwodowych, tj. wątroby, trzustki czy też tkanki tłuszczowej, pośrednio przekłada się na dostępność osoczową substratów energetycznych i wpływa na metabolizm mięśnia sercowego. Niewiele jest bezpośrednich dowodów opisujących skutki aktywacji układu endokannabinoidowego w warunkach fizjologicznych w układzie krążenia. W pracy opisano wpływ chorób metabolicznych, m.in. otyłości i cukrzycy, a także chorób układu krążenia – nadciśnienia, niedokrwienia i zawału na deregulację układu endokannabinoidowego z następczym wpływem na metabolizm kardiomiocytów.
Układ endokannabinoidowy
Układ endokannabinoidowy jest endogennym, kompleksowym systemem sygnałowym biorącym udział w licznych procesach fizjologicznych. Układ ten tworzą:
• receptory endokannabinoidowe (CB – cannabinoid receptor) z rodziny receptorów 7-transbłonowych,
• endokannabinoidy, czyli endogenne ligandy receptorów CB,
• enzymy zaangażowane w syntezę, wychwyt i degradację endokannabinoidów [23,43].
Dotychczas najwięcej informacji o układzie endokannabinoidowym pochodzi z badań nad ośrodkowym układem nerwowym [33].
Receptory endokannabinoidowe
Dotąd odkryto dwa główne rodzaje receptorów endokannabinoidowych – receptor typu I – CB1 oraz receptor typu II – CB2 [48,90]. Zarówno receptor CB1 jak i CB2 należą do grupy receptorów metabotropowych sprzężonych z biał- kiem G i wykazują homologię w 44% [48].
Receptor CB1 jest zbudowany z 472 aminokwasów i jest kodowany przez gen CNR1 [13,61]. Receptory CB1 są umiejscowione głównie na zakończeniach presynaptycznych neuronów ośrodkowego i obwodowego układu nerwowego. Ponadto w mózgu wykryto receptor CB1A, który jest białkowym produktem genu kodującego receptor CB1, powstałym w wyniku jego alternatywnego składania [48]. Należy podkreślić, że receptory CB1 poza ośrodkowym układem nerwowym wykazują ekspresję w licznych narządach obwodowych, tj. mięśniu sercowym, mięśniach szkieletowych, płucach, jądrach, nasieniowodach, macicy, pęcherzu moczowym, nabłonku jelita cienkiego i grubego, a także w tkance tłuszczowej [7,31,81]. Z dotychczas przeprowadzonych badań wynika, że aktywacja receptorów CB1 występujących w mięśniu sercowym wywołuje skutek inotropowy ujemny, natomiast aktywacja receptorów CB1 w obrębie naczyń wieńcowych powoduje ich wazodylatację [7,102]. Ponadto ekspresję receptorów CB1 wykryto w hodowlach komórek śródbłonka tętnic wień- cowych, a także w kardiomiocytach [73,88]. Natomiast w badaniach in vivo stwierdzono obecność receptorów CB1 na zakończeniach presynaptycznych włókien współ- czulnych unerwiających naczynia oporowe i serce oraz na zakończeniach postsynaptycznych włókien współczulnych w naczyniach krwionośnych [62,73,88,89,91]. W badaniach przeprowadzonych na szczurach wykazano, że aktywacja receptorów CB1 występujących na zakończeniach presynaptycznych włókien współczulnych naczyń oporowych i wieńcowych hamuje sekrecję noradrenaliny z tych zakończeń, co prowadzi do zmniejszenia neurogennej odpowiedzi presyjnej i tachykardii [62,91].
Receptor CB2 jest utworzony z 360 aminokwasów, a za jego ekspresję odpowiada gen CNR2 [13]. Ekspresję receptora CB2 wykazano głównie na komórkach i narzą- dach wchodzących w skład układu immunologicznego, a także w strukturach ośrodkowego układu nerwowego, w tym komórkach mikrogleju [99]. Stwierdzono również obecność receptorów CB2 w kardiomiocytach, kardiomioblastach, a także w komórkach śródbłonka tętnic wieńcowych i komórkach mięśni gładkich naczyń krwionośnych [73,86,87,94]. Uważa się, że receptory CB2 są zaangażowane głównie w regulację procesów odpowiedzi immunologicznej [82]. Wykazano, że aktywacja receptorów CB2 w komórkach śródbłonka naczyń wieńcowych zmniejsza wytwarzanie cytokin przeciwzapalnych, w tym czynnika martwicy guza α (TNF-α – tumor necrosis factor alfa) [86]. Tym samym, wpływa to na chemotaksję i adhezję komórek układu odpornościowego do zaktywowanego śródbłonka [86]. Natomiast aktywacja receptora CB2 w komórkach mięśni gładkich tętnic wieńcowych powoduje zahamowanie ich proliferacji [87].
Wyniki wielu prac z ostatnich lat wykazują istnienie jeszcze innych rodzajów receptorów regulujących homeostazę ustroju z udziałem układu endokannabinoidowego. W danych literaturowych dość często wspominana jest obecność tzw. hipotetycznego receptora śródbłonkowego [19,51,89]. Potwierdzeniem powyższego stwierdzenia wydaje się występowanie rozkurczu izolowanej tętnicy krezkowej i towarzyszący temu spadek ciśnienia krwi pod wpływem anandamidu zarówno u myszy transgenicznych CB1-/-/CB2-/-, jak i u myszy niemodyfikowanych genetycznie [51]. Ponadto zniszczenie śródbłonka naczyniowego i podanie rimonabantu (antagonisty receptorów CB1) w stężeniu wyższym niż jest to wymagane do zablokowania receptorów CB1 zniosło opisaną wyżej zarówno wazodylatację, jak i hipotensję [41,51].
Ligandy receptorów endokannabinoidowych
Endogenne ligandy receptorów endokannabinoidowych są pochodnymi kwasu arachidonowego, będącego metabolitem fosfolipidów błon komórek nerwowych [23]. Wykazano, że endokannabinoidy nie są magazynowane, a ich synteza odbywa się natychmiast po zadziałaniu bodźca w postaci potencjału czynnościowego, jest to tzw. „synteza na żądanie” [27]. Uwolnienie endokannabinoidów do przestrzeni synaptycznej następuje podczas depolaryzacji błony komórkowej elementu postsynaptycznego neuronu w wyniku następczego napływu jonów wapnia do wnętrza komórki [14]. Stwierdzono też, że endokannabinoidy należą do transmiterów wstecznych, tzn. są uwalniane z zakończenia postsynaptycznego, a wpływają na element presynaptyczny, gdzie są obecne ich receptory [4,28]. Mechanizm komunikacji między neuronami nazwano indukowanym depolaryzacją zmniejszaniem hamowania (DSI – depolarization-induced suppression of inhibition) lub indukowanym depolaryzacją zmniejszaniem pobudzania (DSE – depolarization-induced suppression of excitation). Zależy to od tego, czy zablokowane zostaje wydzielanie neuromediatorów hamujących, np. kwasu γ-aminomasłowego, czy też pobudzających, np. noradrenaliny, czy też serotoniny z elementu presynaptycznego neuronu [75].
Jak dotąd udało się zidentyfikować cztery związki wykazujące działanie agonistyczne w stosunku do receptorów endokannabinoidowych. Pierwszym z odkrytych endokannabinoidów był anandamid (AEA), który wyizolowano z mózgu świni [21]. Wykazuje większe powinowactwo do receptora CB1 (którego jest częściowym agonistą) ani- żeli CB2 [90]. Anandamid pobudza również receptory N- -metylo-D-asparaginowe (receptory NMDA) [48]. Innym z odkrytych agonistów był 2-arachidonyloglicerol (2-AG), który ma mniejsze powinowactwo do receptora CB1 w porównaniu z anandamidem, a jednocześnie jest pełnym agonistą tego receptora, co oznacza, że po związaniu z receptorem CB1 wywołuje większy efekt niż anandamid. Zarówno w badaniach in vivo, jak i in vitro, wykazano, że oba związki charakteryzują się małą stabilnością wynikająca z budowy chemicznej i szybko ulegają hydrolizie [90]. Innym ligandem w układzie endokannabinoidowym jest eter noladyny, mający podobnie jak 2-AG niskie powinowactwo do receptora CB1. Jednak eter noladyny ze względu na obecność wiązania eterowego ma większą stabilność w porównaniu z poprzednimi agonistami, co może wpływać na wydłużenie wywoływanego przez ten związek działania biologicznego [40]. Do grupy agonistów receptorów endokannabinoidowych zalicza się również endovanilloid (zwany też N-arachidonoyletanolaminą, dopaminą N-arachidonylową, NADA), będący agonistą zarówno receptora CB1, jak też receptora waniloidowego typu 1 (TRPV1) [25]. Istnieją także inne związki wykazujące aktywność wobec receptorów endokannabinoidowych, m.in. palmitoiloetanolamid (PEA), oleiloetanolamid (OEA), będące odpowiednio pochodnymi kwasu palmitynowego i kwasu oleinowego [8,54]. Palmitoiloetanolamid może aktywować receptor CB1, a także CB2 [8]. Natomiast oleiloetanolamid wykazuje niewielkie powinowactwo wobec receptora CB1, przy czym w ogóle nie wpływa na receptory CB2 [54,76].
Jedynym zbadanym endogennym ligandem, który wykazuje cechy antagonistyczne wobec receptorów endokannabinoidowych jest wirodhamina [83]. Synteza tego związku jest związana z estryfikacją kwasu arachidonowego przez etanolaminę. Należy podkreślić, że wirodhamina wykazuje właściwości antagonistyczne tylko w stosunku do receptora CB1, podczas gdy wobec receptora CB2 działa agonistycznie [83]. Poza aktywacją receptorów metabotropowych w komórkach docelowych, endokannabinoidy mają zdolność bezpośredniej aktywacji receptorów jonotropowych, m.in. receptorów TRPV1, serotoninowych 5HT3 oraz nikotynowych dla acetylocholiny z podjednostką α7 [79].
Enzymy i białka transportowe biorące udział w metabolizmie endokannabinoidów
Zgodnie z obecną wiedzą, endokannabinoidy po wydzieleniu do przestrzeni synaptycznej ulegają wychwytowi zwrotnemu. Jest to możliwe dzięki obecności białek transportujących endokannabinoidy, znajdujących się w błonie elementu postsynaptycznego. Najlepiej poznano białko swoiste dla anandamidu zaangażowane w jego wychwyt zwrotny i wewnątrzkomórkowy transport, które nazwano FAAH-podobnym transporterem anandamidu (FLAT – FAAH-like anandamide transporter) [34,64]. Wśród innych białek, które najprawdopodobniej mogą być związane z transportem wewnątrzkomórkowym endokannabinoidów wyróżnić należy: białko wiążące kwasy tłuszczowe 5 i 7 (FABP 5 and 7 – fatty acid binding protein 5 and 7), białko szoku cieplnego 70 (HSP 70 – heat shock protein 70) oraz albuminy [5,52,78]. Wychwycone przez białka transportowe endokannabinoidy pod wpływem enzymu hydrolazy amidowej kwasów tłuszczowych (FAAH) oraz lipazy monoacyloglicerolowej (MGL) we wnętrzu elementu postsynaptycznego ulegają następnie hydrolizie [45]. Stwierdzono, że w stosunku do AEA największą swoistość wykazuje FAAH, natomiast wobec 2-AG zarówno FAAH, jak i MGL są tak samo swoiste [12,45].
Po raz pierwszy MGL jako hydrolazę serynową inaktywującą 2-AG opisano na podstawie doświadczenia przeprowadzonego na mózgu szczura [29]. Powstający w wyniku hydrolizy kwas arachidonowy, może się stać substratem do syntezy m.in. prostaglandyn, co wskazuje, iż prawdopodobnie inhibicja MGL niesie za sobą ogromny potencjał przeciwbólowy i przeciwzapalny [75]. W badaniach przeprowadzonych na szczurach, którym indukowano ból stwierdzono, że zablokowanie rozkładu 2-AG przez inhibitory MGL wywołuje efekt antynocyceptywny zależny od aktywacji receptorów CB1 bądź CB2 [39]. Jednocześnie w innych badaniach wykonanych na myszach, z wyindukowanym zapaleniem jelit, dowiedziono, że po zastosowaniu inhibitorów MGL następuje zmniejszenie ekspresji cytokin prozapalnych, tj. IL-1β i TNF-α [1]. Należy podkre- ślić, że obecność enzymu MGL wykazano również w sercu szczura [53]. Mimo braku badań, nie jest wykluczone, że zastosowanie inhibitorów MGL może mieć znaczenie w zmniejszeniu syntezy prostaglandyn i ograniczeniu rozwoju stanu zapalnego podczas epizodów niedokrwienia mięśnia sercowego.
Znacznie więcej doniesień dotyczy roli FAAH w regulacji układu endokannabinoidowego [16,74,80]. Wyróżnia się dwie izoformy enzymu FAAH – FAAH-1 oraz FAAH-2. Enzym FAAH-1 wykazuje ekspresję u wszystkich ssaków, podczas gdy FAAH-2, nie przejawia ekspresji u wszystkich gatunków, m.in. nie występuje u szczurów [25]. Wewnątrzkomórkowa hydrolityczna aktywność tych enzymów pozwala utrzymać dokomórkowy gradient stężeń endokannabinoidów na odpowiednim poziomie [5]. Najprawdopodobniej enzym FAAH wpływa na możliwości modulowania sygnału przekazywanego przez układ endokannabinoidowy, umożliwiając zastosowanie FAAH w terapii bólu, nudności, a także przeciwdziałaniu uszkodzeniom serca powstałym np. na wskutek przyjmowania leków kardiotoksycznych (m.in. doksorubicyny) [74]. Wykazano, że u myszy pozbawionych genu kodującego FAAH (FAAH-/-) wzrosta poziom anandamidu w miokardium w porównaniu do myszy szczepu dzikiego (FAAH+/+) [80]. Ponadto myszy FAAH-/- w porównaniu do myszy nieznokautowanych wykazują większą wrażliwość na dzia- łanie depresyjne egzogennego anandamidu w układzie sercowo-naczyniowym. Potwierdzeniem obserwacji jest wydłużenie czasu hipotensji zależnej od aktywacji receptorów CB1 oraz zmniejszenie kurczliwości mięśnia sercowego w trójfazowej odpowiedzi hemodynamicznej na dożylne podanie anandamidu u myszy FAAH-/- [80]. Prawdopodobnie jest to wynikiem braku szybkiej inaktywacji egzogennie podanego anandamidu przez różne tkanki [16,80], w tym miokardium [80]. Jednocześnie myszy FAAH-/- zachowują prawidłową czynność serca, wartość ciśnienia krwi oraz wrażliwość baroreceptorów mimo wzrostu poziomu endogennego anandamidu w mięśniu sercowym tych zwierząt [80].
Wiedza na temat enzymatycznej regulacji układu endokannabinoidowego ulega stałemu pogłębianiu. Istnieją doniesienia na temat licznych enzymów, m.in. lipooksygenazy, cytochromu P450, cyklooksygenazy 2, które mogą przekształcać endokannabinoidy do bioaktywnych produktów (pochodnych eikozanoidów, np. etanolamidu prostaglandyny E2), których fizjologiczna rola pozostaje jak dotąd niewyjaśniona [17,103]. Zmiany stężeń zarówno transporterów jak i enzymów mogą zaburzać metabolizm i aktywność endokannabinoidów wytwarzanych w tkankach [45].
Mechanizm aktywacji postreceptorowej w układzie endokannabinoidowym
Pobudzenie receptorów CB1 i CB2 przez swoiste ligandy powoduje aktywację białka Gi/o [48], co w obu przypadkach wywołuje spadek aktywności cyklazy adenylowej i wewnątrzkomórkowego stężenia cAMP [99]. W przypadku receptorów CB1 sugeruje się, że aktywacja białka Gi/o pobudza kaskady kinaz aktywowanych mitogenami (MAPK), odbywającej się przez aktywację kinazy 3-fosfatyloinozytolu [46,48]. Receptory CB1 poprzez aktywację białka Gi/o wpływają na regulację czynności błonowych kanałów jonowych wapnia (kanały typu N, L, P/Q) i potasu (kanałyypu A i Ir). W wyniku aktywacji receptorów CB1 obecne w błonie komórkowej komórek docelowych kanały wapniowe ulegają zamknięciu, przy czym nastę- puje aktywacja i otwarcie kanałów potasowych [48]. Aktualny stan wiedzy wskazuje, że w przypadku aktywacji receptorów CB2 w szlaku transmisji sygnału bierze udział kinaza białkowa C [46] i jak dotąd nie stwierdzono, aby aktywacja receptorów CB2 wpływała na działanie kana- łów jonowych [98].
Ponadto obecne w błonie komórkowej receptory CB1 są związane ze specjalnymi mikrodomenami błonowymi, zwanymi „tratwami lipidowymi” (LR – lipid rafts), które zawierają duże stężenia cholesterolu i glikosfingolipidów. Tratwy lipidowe modulują zdolność wiązania ligandów z receptorami CB1 i wpływają na wewnątrzkomórkowe CB1-zależne szlaki sygnalizacyjne [3]. W badaniach na hodowlach C6 komórek glejaka stwierdzono, iż błonowa zawartość cholesterolu wpływa na integralność tratw lipidowych z błoną komórkową i moduluje tym samym interakcję endokannabinoidów z receptorami [3]. W tych badaniach, w wyniku usuwania cholesterolu z błony komórkowej przez metylo-β-cyklodekstrynę, zauważono zależne od dawki metylo-β-cyklodekstryny zwiększenie wiązania receptorów CB1 ze swoistymi ligandami, m.in. [3H]CP55.940 [3]. Ponadto nastąpił też wzrost przekazywania sygnału zależnego od białek Gi/o związany ze zwiększeniem aktywacji szlaków sygnałowych cyklazy adenylowej i MAPK [3].
Specyfika metabolizmu mięśnia sercowego
W warunkach fizjologicznych w mięśniu sercowym zachodzi metabolizm tlenowy, którego zadaniem jest ciągła resynteza ATP. W celu pozyskania energii są wykorzystywane w 60-70% długołańcuchowe kwasy tłuszczowe (LCFA), w 20-30%, glukoza, a także mleczany i ciała ketonowe [36,96]. Wykazano, że wybór substratu energetycznego zależy od jego dostępności we krwi krążącej, a także od zmian zapotrzebowania energetycznego mię- śnia sercowego [96].
Ze względu na to, iż LCFA stanowią jeden z podstawowych substratów energetycznych mięśnia sercowego, przeprowadzono wiele badań, które dostarczyły istotnych informacji o ich dokomórkowym transporcie. Wykazano, że LCFA dostają się do wnętrza kardiomiocytów na zasadzie biernej dyfuzji (20%) [6] lub z udziałem swoistych białek transportujących kwasy tłuszczowe w procesie transportu wspomaganego (80%) [100]. Do białek tych w mięśniu sercowym zalicza się: FAT/CD36 – translokazę kwasów tłuszczowych, FABPpm – błonowe białko wiążące kwasy tłuszczowe oraz FATP-1, FATP-4 i FATP-6 – białka transportujące kwasy tłuszczowe typu 1, 4 i 6 [30,36]. Ustalono, że białka te znajdują się nie tylko w błonie kardiomiocytów, ale również tworzą pulę zgromadzoną w pęcherzykach wewnątrzkomórkowych mającą zdolność translokacji na powierzchnię błony komórkowej [96]. Translokacja białek transportujących LCFA do błony komórkowej, m.in. pod wpływem czynności skurczowej kardiomiocytów [92], czy też w wyniku stymulacji hormonalnej, np. insuliną [59] zwiększa napływ LCFA do wnętrza kardiomiocytów [59]. W ten sposób możliwe jest dostosowanie podaży substratów energetycznych do aktualnego zapotrzebowania miokardium na energię [6,59]. LCFA po wejściu do wnętrza kardiomiocytów są wiązane przez cytoplazmatyczne białko wiążące kwasy tłuszczowe typu sercowego (H-FABPc) [6]. W takiej postaci LCFA mogą podlegać aktywacji enzymatycznej przez syntetazę acylo-CoA (FACS) po związaniu z koenzymem A (CoA). Aktywny kompleks enzym-substrat łączy się z białkami wiążącymi acylo-CoA (ACBP), co powoduje przemieszczenie LCFA-CoA na dalszy tor przemian w kierunku β-oksydacji lub estryfikacji do określonych frakcji lipidowych [96]. W pierwszym przypadku LCFA związane z CoA dostają się do wnętrza mitochondrium z udziałem enzymu umiejscowionego na zewnętrznej błonie mitochondrialnej – palmitoilotransferazie karnitynowej I (CPT I) oraz karnitynie [58]. Dochodzi do wytworzenia acylokarnityny i uwolnienia CoA. Transport między błoną zewnętrzną a wewnętrzną mitochondrium odbywa się z udziałem kolejnego enzymu, którym jest acylotranslokaza karnitynowa (CAT). Aby doszło do utleniania kwasów tłuszczowych reszta acylowa musi ulec odłączeniu od karnityny i połączyć się z CoA znajdującym się wewnątrz mitochondrium. Na tym etapie katalizowana jest palmitoilotransferaza karnitynowa II. Gdy zostaje odtworzony acylo-CoA, w macierzy mitochondrialnej następuje utlenianie kwasów tłuszczowych, mające na celu wytworzenie energii w postaci ATP, odbywające się dwuetapowo. Pierwszym etapem jest β-oksydacja, podczas której zostaje wytworzony aktywny octan, a następnie jest włączany w cykl kwasów trójkarboksylowych (etap drugi) z wytworzeniem produktów końcowych – dwutlenku węgla oraz wody [18,58]. Zaktywowane LCFA w mniejszym stopniu (10-30%) mogą również tworzyć wewnątrzkomórkową pulę kwasów tłuszczowych z udziałem acylotransferazy glicerolofosforanowej, tworząc wewnątrzkomórkowe zasoby mono-, di- i triacylogliceroli, uwalnianianych w razie zaistnienia zapotrzebowania na LCFA [96].
Jak już wspomniano, drugim pod względem wielkości substratem energetycznym wykorzystywanym przez mię- sień sercowy jest glukoza. Wykazano, że transport glukozy do wnętrza kardiomiocytów odbywa się z udziałem błonowych transporterów GLUT. W metabolizm glukozy w mięśniu sercowym są zaangażowane transportery GLUT1 (przeważając w okresie płodowym) i GLUT4 [18,32]. Transportery GLUT1 są umiejscowione głównie w błonie komórkowej i nie wykazują wrażliwości na stymulację insuliną [32]. Natomiast transportery GLUT4 znajdują się głównie wewnątrz komórki w pęcherzykach mikrosomalnych. Translokacja GLUT4 na powierzchnię błony komórkowej zachodzi podczas stymulacji przez róż- ne czynniki, wśród których wymienić należy insulinę, niedokrwienie oraz przeciążenie serca (overload) [32]. W warunkach fizjologicznych w sercu występuje również niska ekspresja transportera GLUT3 [20]. Oprócz wymienionego transportu istnieje również kotransport sodu i glukozy z udziałem transportera SGLT1, reagującego na stymulację insuliną oraz leptyną [2]. Glukoza po wejściu do wnętrza komórki w wyniku procesu glikolizy przekształcana zostaje w pirogronian. W warunkach tlenowych pirogronian pod wpływem enzymu dehydrogenazy pirogronianowej (PDH) przekształca się do acetylo-CoA, który jest włączany do cyklu kwasów trójkarboksylowych (Krebsa). Przy braku dostępności tlenu dehydrogenaza pirogronianowa (LDH) powoduje powstawanie mleczanu z pirogronianu, jednak należy podkreślić, iż procesy glikolizy beztlenowej w mięśniu sercowym są znikome [18].
Wpływ endokannabinoidów na metabolizm mięśnia sercowego
Pobór substratów energetycznych przez mięsień sercowy zależy od wielu czynników, wśród których wymienić należy: stężenie docierających do kardiomiocytów substratów, wpływ hormonów regulujących metabolizm (m.in. insulina), zapotrzebowanie serca na energię (wysiłek fizyczny, tachykardia, wzrost obciążenia następczego) oraz zaopatrzenie miokardium w tlen [96]. Pośrednio lub bezpośrednio każdy czynnik może aktywować układ endokannabinoidowy. Brak jest danych literaturowych opisujących bezpośredni wpływ endokannabinoidów na metabolizm miokardium. Ponieważ receptory tego układu są umiejscowione nie tylko na powierzchni kardiomiocytów, ale również występują na zakończeniach włókien współczulnych autonomicznego układu nerwowego unerwiającego serce, z tego można wnioskować, iż endokannabinoidy są zaangażowane w metabolizm mięśnia sercowego w sposób bezpośredni. Możliwe jest również, że endokannabinoidy wykazują wpływ pośredni, modulując transmisję neuronalną przez umiejscowione w ośrodkowym i obwodowym układzie nerwowym komponenty tego układu, wpływając na podwzgórzową regulację poboru pokarmu, metabolizm substratów energetycznych w wątrobie i ostatecznie zmieniając przepływ wieńcowy [15,48].
Pośredni wpływ endokannabinoidów na metabolizm mięśnia sercowego
Wiele danych doświadczalnych dowodzi wpływu układu endokannabinoidowego na wzmożone łaknienie [9,15,35,49]. Działanie to jest możliwe przez wpływ na receptory CB1 obecne w obrębie podwzgórza (jądro przykomorowe i jądro łukowate podwzgórza). W badaniach przeprowadzonych na szczurach udowodniono, że przez bezpośrednią iniekcję anandamidu do podwgórza można wywołać uczucie głodu [49]. Ponadto endokannabinoidy aktywując receptory CB1 w podwgórzu działają na uwalnianie peptydów oreksygenicznych regulujących pobieranie pokarmu, np. neuropeptyd Y, hormon melanocytotropowy czy preprooreksynę 1. Ekspresję receptorów CB1 stwierdzono również w neuronach podwzgórza wydzielających peptydy anoreksygeniczne, czyli zmniejszające łaknienie, m.in. kortykoliberynę, transkrypt regulowany przez kokainę i amfetaminę. Prawdopodobnie przez aktywację receptorów CB1 jest możliwe wpływanie na ich wydzielanie i tym samym regulację apetytu [15,35,49].
Aktywacja receptorów CB1 w obrębie układu mezolimbicznego reguluje nie tylko łaknienie, ale bierze też udział w procesach motywacji i aktywacji behawioralnej w wyniku zadziałania czynnika nagradzającego (konsumpcji pokarmu) [9,15]. W badaniu przeprowadzonym na zdrowych ochotnikach wykazano, że konsumpcji ulubionych potraw w porównaniu do potraw o neutralnych walorach smakowych towarzyszy wzrost stężenia 2-arachidonyloglicerolu w osoczu, który pozytywnie koreluje z osoczowym stężeniem greliny [71]. Wynik eksperymentu może świadczyć o prawdopodobnym zaangażowaniu aktywacji układu endokannabinoidowego w występowanie hiperfagii hedonicznej, czyli spożywania pokarmów dla przyjemności, a nie w celu utrzymania homeostazy energetycznej [71]. Nadmierna podaż pokarmu przyczynia się do wzrostu dostępności substratów energetycznych w osoczu, co istotnie wpływa na energetykę mięśnia sercowego, ponieważ duże stężenie krążących kwasów tłuszczowych bezpośrednio wzmaga dokomórkowy napływ LCFA do kardiomiocytów. Mimo iż kwasy tłuszczowe są podstawowym źródłem energii dla miokardium to wzrost ich metabolizowania wywiera negatywne skutki. Dzieje się tak dlatego, że wraz ze wzrostem oksydacji kwasów tłuszczowych jako substratu energetycznego maleje udział glukozy (a także mleczanów) w przemianach metabolicznych kardiomiocytów. Powyższa zmiana w metabolizmie miokardium zmniejsza wydolność mechaniczną serca, a także zwiększa ilość zu- żywanego tlenu do wygenerowania ATP niezbędnego do skurczu [96]. Ponadto w doświadczeniu przeprowadzonym na myszach wykazano, że nawet podczas 3-dniowego zastosowania diety bogatej w tłuszcze dochodzi do przejściowego wzrostu poziomu 2-arachidonyloglicerolu w podwzgórzu [44]. Natomiast w przypadku wydłużenia okresu stosowania tej diety do 14 dni, stwierdzono trwały wzrost 2-arachidonyloglicerolu w podwzgórzu [44]. W obydwu opisanych doświadczeniach zauważono zwiększenie preferencji do spożywania pokarmów obfitujących w tłuszcze, ustępujące po zastosowaniu antagonisty receptora CB1. Informacje te wskazują, że w wyniku nadmiernej aktywacji receptorów CB1 pod wpływem podwyższonego stężenia 2-arachidonyloglicerolu może wystąpić skłonność do konsumpcji pokarmów wysokotłuszczowych [44]. W wyniku spożywania zwiększonej ilości tłuszczów w diecie wzrasta stężenie krążących wolnych kwasów tłuszczowych, co w rezultacie wpłynie na zwiększenie dostępności tego substratu energetycznego dla kardiomiocytów. Di Marzo i wsp. [26] w badaniach na zwierzętach wykazali ponadto, że w podwzgórzu występuje pewien rodzaj sprzężenia zwrotnego między 2-AG a leptyną. Zauważono bowiem, że podwyższonemu stężeniu 2-AG towarzyszy upośledzenie sygnału przekazywanego przez leptynę u otyłych myszy db/db i ob/ob i szczurów Zucker [26]. Natomiast jednorazowe podanie leptyny redukuje stężenie 2-AG u myszy ob/ob i szczurów bez mutacji genetycznej [26]. Powyż- sze odkrycie wskazuje na udział 2-AG w podwzgórzu jako komponenty wchodzącej w skład obwodów neuronalnych regulowanych przez leptynę. Ponadto toniczne pobudzanie receptorów CB1 przez 2-AG najprawdopodobniej ma na celu podtrzymanie pobierania pokarmu [26]. Sugeruje się, że podwyższone stężenie 2-AG i towarzyszące mu zaburzenia w szlaku leptynowym [26] mogą mieć udział w obwodowych zaburzeniach metabolicznych, np. w wą- trobowej oporności na insulinę [77]. Wykazano, że aktywacja receptorów CB1 w obrębie ośrodkowego układu nerwowego u szczurów z infuzją agonistów receptorów CB1 – WIN55,212-2 (WIN) lub arachidonoilo-2-chloroetylamidu (ACEA) do komór mózgu hamuje zależną od insuliny supresję wątrobowego wytwarzania glukozy, co powoduje nadmierne wytwarzanie glukozy przez wątrobę [77]. Zaobserwowane zahamowanie supresji wątrobowej wytwarzanej glukozy nie wynika ze zmian w wewnątrzkomórkowym szlaku insulinowym w wątrobie, ponieważ zarówno ekspresja, jak i fosforylacja istotnych składowych tego szlaku – kinazy białkowej AKT oraz kinazy syntazy glikogenu 3 – GSK3 w wątrobie nie zmieniły się pod wpływem WIN i ACEA [77]. Najprawdopodobniej zahamowanie zależnego od insuliny, wątrobowego wytwarzania glukozy pod wpływem WIN i ACEA jest wynikiem zaburzeń w szlaku insulinowym i leptynowym na poziomie ośrodkowego układu nerwowego, przy czym mechanizm ten nie został wyjaśniony [56]. Ponadto w wyniku centralnej aktywacji receptorów CB1 upośledzona zostaje zdolność insuliny do hamowania ogólnoustrojowej lipolizy białej tkanki tłuszczowej (WAT – white adipose tissue) wskutek zwiększenia ekspresji i fosforylacji białka przenoszącego lipidy – perilipiny oraz koaktywatora lipazy triacyloglicerolowej (ATGL – adipose triglyceride lipase) – CGI-58 (comparative gene identification-58) [77]. Dochodzi także do wzrostu ekspresji głównych enzymów lipolitycznych w WAT – ATGL i lipazy hormonowrażliwej (HSL – hormone-sensitive lipase). Uwolniony z WAT glicerol może stanowić podłoże do glukoneogenezy w wątrobie, zwiększając tym samym stężenie glukozy we krwi i mechanizm obserwowanej w wątrobie oporności na insulinę [77]. Zwiększona lipoliza przyczynia się do podwyższenia stężenia krążących wolnych kwasów tłuszczowych, a przez to do wygenerowania lipotoksyczności, która jest jednym z czynników patogenetycznych w rozwoju cukrzycy typu 2. Powyższe zmiany wpływają na zwiększenie dostępności substratów energetycznych dla wszystkich komórek, w tym również dla kardiomiocytów. Zwiększenie osoczowej biodostępności glukozy i kwasów tłuszczowych zwiększa dokomórkowy transport tych substratów energetycznych do wnętrza kardiomiocytów [26,77,93].
Ze względu na obecność receptorów endokannabinoidowych w obrębie tkanek i narządów obwodowych o znacznej aktywności metabolicznej, endokannabinoidy wpływają na gospodarkę energetyczną ustroju także poza ośrodkowym układem nerwowym. Wykazano, że u myszy szczepu dzikiego obecne w dojrzałych adipocytach receptory CB1 w wyniku aktywacji w warunkach in vitro powodują zwiększenie syntezy kwasów tłuszczowych de novo, wewnątrzkomórkową akumulację triglicerydów oraz zmniejszenie lipolizy [101]. Podłożem zmian jest pobudzenie wychwytu glukozy i aktywacja enzymu lipogenezy, tj. syntazy kwasów tłuszczowych (FAS – fatty acid synthase). Prawdopodobnie prowadzi to do zmniejszenia stężenia wolnych kwasów tłuszczowych i glukozy we krwi. Ponadto, w wyniku aktywacji receptora CB1 dochodzi do zahamowania śródbłonkowej syntazy tlenku azotu i następczego spadku biogenezy i aktywności enzymów mitochondrialnych w adipocytach [101].
Stwierdzono, że receptory CB1 znajdujące się na zakończeniach nerwowych przewodu pokarmowego pod wpływem anandamidu stymulują łaknienie, wpływając tym samym na zwiększenie pobierania pokarmu i wzrost dostępnych w ustroju substratów energetycznych [15]. Przypuszcza się, że endokannabinoidy hamują opróż- nianie żołądka oraz zmniejszają perystaltykę jelit, co również sprzyja zwiększonemu wchłanianiu substratów energetycznych do krwiobiegu. Obecność receptorów CB1 w wątrobie, mimo iż występują w niewielkiej ilości, wpływa na możliwość regulowania metabolizmu glukozy przez endokannabinoidy. Wykazano, że systemowa blokada receptorów CB1 przez antagonistę AVE1625 u szczurów wywołuje wzrost wątrobowej glikogenolizy, co świadczy o prawdopodobnym udziale aktywacji receptorów CB1 w tworzeniu glikogenu w wątrobie [42]. Być może centralna aktywacja receptorów CB1 ma na celu dostarczenie substratów energetycznych dla organizmu, natomiast aktywacja obwodowych receptorów CB1 sprzyja efektywnemu gromadzeniu pozyskanych źródeł energii [24].
Przez obecność receptorów CB1 i CB2 w komórkach β trzustki endokannabinoidy mogą wpływać na metabolizm regulując wydzielanie insuliny. Nie jest jednak pewne, czy aktywacja receptorów CB1 kontroluje tylko sygnał przekazywany przez insulinę czy wpływa na jej wydzielanie [95]. Liu i wsp. [56] w badaniach przeprowadzonych na ludzkich i mysich hepatocytach sugerują, że aktywacja receptorów CB1 pod wpływem anandamidu powoduje zahamowanie sygnału przekazywanego przez insulinę w dwojaki sposób. Indukuje fosforylację seryny w pozycji 307 substratu receptora insuliny typu 1 (IRS1 – insulin receptor substrate 1), co wpływa na zahamowanie aktywności kinazy białkowej B (AKT2). Aktywacja receptorów CB1 wpływa bezpośrednio na zahamowanie indukowanej insuliną fosforylacji AKT2. Kinaza AKT2 będąc w postaci nieufosforylowanej jest nieaktywna i tym samym hamowany jest szlak insulinowy [56]. Główną funkcją AKT2 jest aktywacja kaskady reakcji prowadzących do zwiększenia translokacji transporterów GLUT4 z puli wewnątrzkomórkowej do błony komórkowej pod wpływem stymulacji insuliną [38]. Oznacza to, że glukoza mimo obecności insuliny nie może zostać przetransportowana do wnętrza hepatocytów [56]. Esposito i wsp. [31] w badaniu przeprowadzonym na hodowli C6 komórek mięśni szkieletowych myszy również zauważyli, że układ endokannabinoidowy wpływa ujemnie na regulację szlaków insulinowych P13K-AKT, a także P13K-PDK-PKCξ, których udział jest niezbędny w wychwycie glukozy z udziałem transporterów GLUT4 [95]. Dowiedziono, że komórki α wysp trzustki wytwarzają enzymy biorące udział w syntezie endokannabinoidów, natomiast komórki β syntetyzują enzymy degradujące endokannabinoidy [97]. Zanim endokannabinoidy ulegną hydrolizie, mogą zaktywować receptory CB2 występujące w komórkach β trzustki i spowodować zahamowanie wydzielania insuliny [97]. Ponadto anandamid działając na receptory waniloidowe TRPV1, które są sprzężone z komórkami β trzustki, również wpływa na ich czynność wydzielniczą [97]. Endokannabinoidy przez zahamowanie szlaków insulinowych, a także translokacji transporterów glukozy prawdopodobnie mogą wpłynąć na zmniejszenie stężenia glukozy wewnątrz kardiomiocytów, a tym samym spadek metabolizmu tego substratu energetycznego w miokardium.
Podsumowując wydaje się, że wpływ aktywacji układu endokanabinoidowego na metabolizm jest zarówno ośrodkowy (ogólnoustrojowy) jak i miejscowy (narządowy) (ryc. 1).
Endokannabinoidy w otyłości
Obecnie wiele badań wskazuje, że rozwojowi otyłości towarzyszy aktywacja układu endokannabinoidowego [11,24,37,57,65,66]. Świadczy o tym m.in. występująca u myszy dysregulacja układu endokannabinoidowego powstała w wyniku otyłości wyindukowanej dietą wysokotłuszczową, która objawia się wczesnym i trwałym wzrostem ekspresji anandamidu i 2-AG w wielu tkankach obwodowych, m.in. w sercu [67]. Ze względu na ich właściwości lipogeniczne i jednoczesny wzrost ekspresji receptorów CB1 w sercu może dojść do wzmożenia lipogenezy [67]. Nadmierna lipogeneza może się przyczynić do stłuszczenia osierdzia, co stanowi jeden z czynników ryzyka chorób sercowo-naczyniowych [24,67]. Przeprowadzono także badania mające na celu stwierdzenie czy możliwa jest supresja stymulowanej przez układ endokannabinoidowy lipogenezy. W badaniach tych wykazano, że długotrwałe zablokowanie receptorów CB1 w WAT przez rimonabant zwiększa utlenianie kwasów tłuszczowych i wydatek energetyczny u myszy z otyłością wywołaną dietą wysokotłuszczową [50]. Prawdopodobnie rimonabant wpływa na zwiększenie ekspresji enzymów w WAT zaangażowanych w utlenianie kwasów tłuszczowych [50]. Przeprowadzono też eksperyment na genetycznym modelu otyłości – u szczurów Zucker, któ- rym także podawano rimonabant [69]. Inhibicja receptorów CB1 przyczyniła się do spadku masy ciała, hamowania procesu akumulacji tkanki tłuszczowej, wzrostu wrażliwości na insulinę, zwiększonej aktywności układu współczulnego, a także wzrostu stężenia adiponektyny oraz zmniejszenia ciśnienia krwi [57,69]. Z powyższych doświadczeń wynika, że skutki nadmiernej aktywacji układu endokannabinoidowego mogą być łagodzone przez antagonistę receptorów CB1 zarówno w otyłości wynikającej z nieprawidłowej diety, jak i otyłości spowodowanej czynnikami genetycznymi.
Niemniej interesującym wydaje się badanie, w którym wykazano, że aktywność enzymu FAAH w dojrzałych adipocytach pozytywnie koreluje ze wzrostem wskaźnika masy ciała (BMI – body mass indeks) i obwodem talii u zdrowych osób [11]. Prawdopodobnie wzrost aktywności enzymu FAAH jest wynikiem ogólnego wzrostu aktywności układu endokanabinoidowego w czasie rozwoju otyłości. Jednocześnie obserwacja potwierdza hipotezę, że niektó- re składowe układu endokannabinoidowego są pobudzane w wyniku pojawienia się chorób, m.in. otyłości [11].
Endokannabinoidy w cukrzycy
Podobnie jak w otyłości, istnieje wiele badań wskazują- cych, że aktywacja receptorów CB1 sprzyja rozwojowi cukrzycy typu 2 [57]. Liu i wsp. w badaniach na myszach zauważyli, że po zastosowaniu ich blokera – rimonabantu nastąpiło zwiększenie wychwytu i metabolizmu glukozy, m.in. w mięśniach szkieletowych, co jednocześnie wskazuje na zależne od aktywacji receptorów CB1 zaburzenie przemian metabolicznych glukozy [57]. W celu wyjaśnienia molekularnego mechanizmu zaburzeń metabolizmu glukozy warto jest zwrócić uwagę na wyniki doświadczenia przeprowadzonego przez Motaghediego i wsp. [72]. Wykazali związek między zastosowaniem antagonisty receptorów CB1 a zwiększeniem translokacji transportera glukozy GLUT4 z kompartmentu wewnątrzkomórkowego do błony komórkowej w adipocytach [72]. Oznacza to, że aktywacja receptorów CB1 hamuje translokację transportera GLUT4, a dane literaturowe wskazują, że opisany mechanizm występuje również m.in. w komórkach mięśni szkieletowych [47], co nie wyklucza udziału hamowania translokacji GLUT4 przez zaktywowane receptory CB1 w kardiomiocytach. W wyniku spadku liczby błonowych transporterów glukozy wychwyt glukozy z krwi do komó- rek ulega zmniejszeniu, prowadząc do spadku intensywności metabolizowania glukozy w komórkach. Ponadto badaniom poddano wpływ układu endokannabinoidowego w rozwoju cukrzycy typu 1 [85]. Rajesh i wsp. dowiedli, że cukrzyca typu 1 doprowadza do wzrostu ekspresji receptorów CB1 i wzrostu stężenia anandamidu w sercu myszy [85]. Ponadto dysfunkcja mięśnia sercowego powstająca w przebiegu cukrzycy ulega poprawie u myszy CB1-/- oraz u myszy nieznokautowanych leczonych antagonistami receptorów CB1 [85]. Genetyczna delecja lub farmakologiczna inhibicja receptorów CB1 osłabia aktywację MAPK, zmniejsza apoptozę komórek, stan zapalny oraz stres oksydacyjny i nitrozacyjny obejmujący miokardium myszy z cukrzycą [85]. Wyniki badań otrzymanych Podobnie jak w otyłości, istnieje wiele badań wskazują- cych, że aktywacja receptorów CB1 sprzyja rozwojowi cukrzycy typu 2 [57]. Liu i wsp. w badaniach na myszach zauważyli, że po zastosowaniu ich blokera – rimonabantu nastąpiło zwiększenie wychwytu i metabolizmu glukozy, m.in. w mięśniach szkieletowych, co jednocześnie wskazuje na zależne od aktywacji receptorów CB1 zaburzenie przemian metabolicznych glukozy [57]. W celu wyjaśnienia molekularnego mechanizmu zaburzeń metabolizmu glukozy warto jest zwrócić uwagę na wyniki doświadczenia przeprowadzonego przez Motaghediego i wsp. [72]. Wykazali związek między zastosowaniem antagonisty receptorów CB1 a zwiększeniem translokacji transportera glukozy GLUT4 z kompartmentu wewnątrzkomórkowego do błony komórkowej w adipocytach [72]. Oznacza to, że aktywacja receptorów CB1 hamuje translokację transportera GLUT4, a dane literaturowe wskazują, że opisany mechanizm występuje również m.in. w komórkach mięśni szkieletowych [47], co nie wyklucza udziału hamowania translokacji GLUT4 przez zaktywowane receptory CB1 w kardiomiocytach. W wyniku spadku liczby błonowych transporterów glukozy wychwyt glukozy z krwi do komó- rek ulega zmniejszeniu, prowadząc do spadku intensywności metabolizowania glukozy w komórkach. Ponadto badaniom poddano wpływ układu endokannabinoidowego w rozwoju cukrzycy typu 1 [85]. Rajesh i wsp. dowiedli, że cukrzyca typu 1 doprowadza do wzrostu ekspresji receptorów CB1 i wzrostu stężenia anandamidu w sercu myszy [85]. Ponadto dysfunkcja mięśnia sercowego powstająca w przebiegu cukrzycy ulega poprawie u myszy CB1-/- oraz u myszy nieznokautowanych leczonych antagonistami receptorów CB1 [85]. Genetyczna delecja lub farmakologiczna inhibicja receptorów CB1 osłabia aktywację MAPK, zmniejsza apoptozę komórek, stan zapalny oraz stres oksydacyjny i nitrozacyjny obejmujący miokardium myszy z cukrzycą [85]. Wyniki badań otrzymanych przez Rejesha i wsp. dotyczące wzrostu ekspresji receptorów CB1 i stężenia anandamidu w lewej komorze serca myszy z cukrzycą typu 2 są zgodne z wynikami wzrostu stężenia anandamidu w ludzkiej siatkówce oka objętej retinopatią cukrzycową [68,85]. Powyższe wyniki dowodzą ogólnoustrojowego wpływu układu endokannabinoidowego na organizm podczas cukrzycy.
Endokannabinoidy a układ krążenia
Endokannabinoidy in vivo mają bezpośredni wpływ na układ sercowo-naczyniowy [17]. Badania przeprowadzone na szczurach dowiodły, że anandamid może wywoływać zmiany hemodynamiczne występujące w postaci trzech faz [63]. W fazie I dochodzi do jednoczesnej aktywacji receptorów TRPV1 i wystąpienia bradykardii oraz hipotensji. W fazie II następuje krótkotrwała odpowiedź presyjna. Sugeruje się, że faza ta ma złożony mechanizm, w którym są angażowane struktury ośrodkowe (receptory ß-adrenergiczne i NMDA) i obwodowe (naczynia krwionośne; mechanizm zależny od kanałów wapniowych). Prawdopodobnie w tej fazie dochodzi również do aktywacji receptorów TRPV1. W fazie III obserwuje się wydłużoną hipotensję, w której udział biorą receptory CB1 znajdujące się w naczyniach krwionośnych oraz w sercu. Ich aktywacja prowadzi do zahamowania zarówno odpowiedzi presyjnej, jak i tachykardii [63]. Ponadto wykazano wpływ metabolitu anandamidu powstałego w wyniku hydrolizy pod wpływem enzymu FAAH na kanały potasu zależne od ATP (KATP) w izolowanych szczurzych kardiomiocytach komór serca [55]. Powstały podczas hydrolizy anandamidu kwas arachidonowy wpływa na aktywację kana- łów KATP w szczurzych kardiomiocytach [55]. Kanały te są odpowiedzialne za metabolizm energetyczny komórek, tzn. reagują na zmiany wewnątrzkomórkowego stosunku ATP/ADP i są zamykane gdy stosunek ten wzrasta. Ich otwarcie następuje pod wpływem obniżenia wewnątrzkomórkowej zawartości ATP oraz spadku ekspresji kana- łów potasu zależnych od acetylocholiny (KAch). Otwarcie kanałów potasowych powoduje wypływ jonów potasu z komórki i następczą repolaryzację błony komórkowej kardiomiocytów. Aktywacja kanałów KATP przez metabolit anandamidu ma znaczenie antyarytmiczne i kardioprotekcyjne podczas niedotlenienia mięśnia sercowego [55]. Otwarcie kanałów KATP skraca czas trwania potencja- łu czynnościowego kardiomiocytów i zmniejsza dopływ wapnia przez kanały typu L, co zapobiega nadmiernemu wzrostowi stężenia wapnia w sercu i utrzymuje ilość ATP w kardiomiocytach na odpowiednim poziomie [55]. Układ endokannabinoidowy nie wykazuje tonicznej aktywności w warunkach fizjologicznych w obrębie układu krążenia. Zauważono natomiast, że w procesach patologicznych kompensuje zmiany w parametrach krążenia, m.in. przez zmiany w wydzielaniu endokannabinoidów i ekspresji receptorów endokannabinoidowych [10,62,63,80].
Endokannabinoidy w nadciśnieniu
Wyniki badań wskazują na wpływ aktywacji receptorów CB1 przez endokannabinoidy na obniżenie ciśnienia tętniczego krwi w nadciśnieniu tętniczym u szczurów [10]. W różnych postaciach nadciśnienia tętniczego (samoistnego, nerkopochodnego) występuje toniczna aktywacja układu endokannabinoidowego za co prawdopodobnie odpowiada zwiększona ekspresja receptorów CB1 w sercu i komórkach endotelialnych naczyń krwionośnych [10]. W doświadczeniu przeprowadzonym na szczurach z nadciśnieniem samoistnym (SHR – spontaneously hypertensive rat) stwierdzono, że zwiększona ekspresja receptorów CB1 w kardiomiocytach i komórkach endotelialnych aorty szczurów SHR w porównaniu do szczurów normotensyjnych przekłada się na wzrost działania hipotensyjnego w odpowiedzi na podanie kannabinoidów pochodzenia egzogennego u szczurów SHR [10]. Ponadto zwiększenie aktywacji receptorów CB1 przez zablokowanie degradacji i wewnątrzkomórkowego wychwytu zwrotnego anandamidu normalizuje ciśnienie krwi, o czym świadczy wpływ inhibicji enzymu FAAH przez URB597 u szczurów SHR na zmniejszenie ciśnienia krwi oraz czynności serca do poziomu obserwowanego u zwierząt normotensyjnych. Dodatkowych informacji na temat zależności występujących między nadciśnieniem a układem endokannabinoidowym dostarczyło zastosowanie antagonisty receptorów CB1 – SR141716 u szczurów SHR. U szczurów z nadciśnieniem samoistnym zastosowanie SR141716 wywołuje trwały dalszy wzrost ciśnienia krwi, co sugeruje że prawdopodobnie podniesione podstawowe ciśnienie krwi wpływa na toniczną aktywację receptorów CB1 [10]. Ponadto antagoniści receptorów CB1 zwiększają wydolność skurczową serca bez znaczącego wpływu na obwodowy opór naczyniowy, co może oznaczać, że endokannabinoidy podczas nadciśnienia działają głównie na serce [10]. Antagoniści receptora CB1 oraz inhibitory FAAH zastosowane u szczurów normotensyjnych nie wywoływały żadnego działania. Wyniki tych badań mogą świadczyć o widocznej aktywacji układu endokannabinoidowego w układzie krążenia w stanie hipertensji, a zastosowanie agonistów receptorów CB1 i inhibitorów agonistów receptorów CB1 przyczynia się do normalizacji ciśnienia tętniczego krwi [10].
Endokannabinoidy w niedokrwieniu i zawale mięśnia sercowego
Z dotychczas przeprowadzonych badań wynika, że endokannabinoidy wykazują właściwości kardioprotekcyjne. W eksperymencie przeprowadzonym na szczurach dowiedziono, że 2-AG i PEA podczas niedokrwienia mięśnia sercowego zmniejszają w istotny sposób obszar uszkodzenia tkanki [60]. Jednocześnie zmniejszeniu uległo stężenie enzymów uwolnionych z kardiomiocytów – kinazy kreatynowej i dehydrogenazy mleczanowej [60]. Ponadto w mysim modelu niedokrwienia i reperfuzji miokardium wykazano, że stężenie troponiny I pochodzą- cej z kardiomiocytów objętych martwicą jest znacznie mniejsze w przypadku zastosowania agonistów receptorów CB2 [70]. Prawdopodobnie aktywacja receptorów CB2 przyczynia się do „hartowania” serca niedokrwieniem (preconditioning), czyli zabezpiecza kardiomiocyty przed nekrozą w wyniku niedokrwienia oraz następczej reperfuzji [60]. W doświadczeniu przeprowadzonym na myszach, u których wyidukowano zawał mięśnia sercowego dowiedziono, że aktywacja receptorów CB2 zmniejsza migrację leukocytów w miejsce uszkodzonej tkanki [22]. Dochodzi do zmniejszenia możliwości zachodzenia interakcji między leukocytami a śródbłonkiem, a tym samym obniżeniu ulega prawdopodobieństwo wystąpienia dodatkowego uszkodzenia przez zaktywowane komórki układu odpornościowego [22]. Wykazano także, że aktywacja receptorów CB2 przyczynia się do zmniejszonego wytwarzania reaktywnych form tlenu przy zmniejszeniu infiltracji neutrofili w obrębie komór serca po zawale [70].
Endokannabinoidy wykazują właściwości kardioprotekcyjne, ale istnieją dowody na ich negatywny wpływ na układ krążenia. Wykazano, że pobudzenie receptorów CB1 przez AEA w ludzkich komórkach śródbłonka tętnic wieńcowych skutkuje zależną od dawki oraz czasu stymulacją aktywacji szlaków p38 i JNK-MAPK, wzrostem wytwarzania reaktywnych form tlenu oraz śmiercią komórek głównie w wyniku apoptozy [88]. Ponieważ zaktywowane receptory CB1 przez stymulację szlaków p38 i JNK-MAPK promują odpowiedź zapalną, a zaktywowane receptory CB2 wpływając na zmniejszenie ekspresji cytokin prozapalnych hamują chemotaksję i aktywację leukocytów tłumiąc reakcje immunologiczne [88] nasuwa się wniosek: w chwili powstania niedokrwienia mię- śnia sercowego w komórkach śródbłonka zaktywowane receptory CB1 i CB2 pełnią przeciwstawną w stosunku do siebie funkcję – receptory CB1 prozapalną, natomiast receptory CB2 przeciwzapalną. Jednocześnie warto zauważyć, że zastosowanie agonisty receptorów CB2 i/lub antagonisty receptorów CB1 może wpłynąć ochronnie na poddane niedokrwieniu kardiomiocyty. Quercioli i wsp. w badaniu dotyczącym określenia zależności między stę- żeniem endokannabinoidów a występowaniem dysfunkcji w krążeniu wieńcowym u ludzi otyłych [84] stwierdzili, że podwyższone stężenie anandamidu i 2-AG w osoczu wykazuje pozytywną korelację ze zmniejszonym przepływem krwi przez miokardium u osób otyłych w porównaniu do osób o prawidłowej masie [84]. Prawdopodobobnie pod wpływem nadmiernej aktywacji układu endokannabinoidowego dochodzi do nieprawidłowości w funkcjonowaniu śródbłonka i komórek mięśni gładkich tętniczek wieńcowych, co przekłada się na zauważone w doświadczeniu zmiany w przepływie krwi przez miokardium [84].
Podsumowanie
Mimo iż niewiele wiadomo o bezpośrednim wpływie układu endokannabinoidowego na metabolizm mięśnia sercowego, to należy przypuszczać, iż przez ogólny wpływ na metabolizm organizmu przyczynia się do zmian w obrębie miokardium. Z tego też względu upatrywać można nowych możliwości terapeutycznych związanych z udziałem endokannabinoidów w leczeniu licznych zaburzeń serca – nadciśnienia, arytmii, niedokrwienia, zawału, w których dochodzi do nieprawidłowości w zaopatrzeniu energetycznym miokardium. Dlatego też lepsze poznanie bezpo- średniego wpływu tego układu pozwoliłoby kontrolować metabolizm mięśnia sercowego i umożliwiłoby przeciwdziałanie zachodzącym zmianom, które w przypadku mię- śnia sercowego mogą mieć tragiczne skutki.
Otyłość trzewna stanowi główny czynnik ryzyka rozwoju insulinooporności, która może prowadzić do jawnej cukrzycy typu 2 (T2D) z utratą funkcji komórek beta i ostateczną utratą tych komórek. Wydzielanie insuliny przez komórki beta wysp trzustkowych jest ściśle związane z stężeniem glukozy we krwi i regulowane przez wiele mediatorów uwalnianych do krwiobiegu lub miejscowo, w tym endokannabinoidów. Otyłość i jej powikłania, w tym T2D, wiążą się z zwiększoną aktywnością układu endokannabinoidowego/receptora CB1 (CB1R), co potwierdzają efekty terapeutyczne antagonistów CB1R. Podobne korzystne efekty antagonistów CB1R o ograniczonej penetracji do mózgu wskazują na istotną rolę CB1R w tkankach obwodowych, w tym trzustce endokrynnej. Komórki beta trzustki wykazują ekspresję wszystkich składników układu endokannabinoidowego, a endokannabinoidy modulują ich funkcję zarówno poprzez mechanizmy autokrynne, jak i parakrynne. Oddziałują one na wydzielanie insuliny zarówno w warunkach bazalnych, jak i indukowanych glukozą, wpływając również na proliferację i przeżycie komórek beta. Niniejsza krótka recenzja przedstawi dostępne informacje na temat modulacji tych procesów przez endokannabinoidy i receptory, starając się ocenić wkład takich efektów w kontrolę glikemii w T2D i insulinooporności.
Cukrzyca typu 2 (T2D) lub cukrzyca nieinsulinoniezależna to przewlekła choroba charakteryzująca się zaburzoną zdolnością organizmu do metabolizowania glukozy, która często rozwija się u osób otyłych/nadwadze. Otyłość jest czynnikiem ryzyka rozwoju insulinooporności, która definiowana jest jako niemożność normalnej odpowiedzi komórek w tkankach wrażliwych na insulinę na insulinę produkowaną i uwalnianą przez komórki beta wysp trzustkowych w odpowiedzi na wzrost stężenia glukozy we krwi. Powszechnie przyjętym poglądem jest, że u podgrupy otyłych, insulinoopornych jednostek dochodzi do dysfunkcji komórek beta, co prowadzi do zmniejszonej produkcji insuliny, złej regulacji glukozy we krwi, a ostatecznie do T2D [1]. Istnieje także możliwość, że dysfunkcja komórek beta występuje przed lub równolegle do insulinooporności, ponieważ w niektórych przypadkach może być wykrywana znacznie przed wystąpieniem T2D [2].
Komórki beta wysp trzustkowych są wyjątkowo wrażliwe na zmiany stężenia glukozy we krwi. Białko przewoźnika glukozy typu 2 (GLUT-2) umożliwia wejście glukozy do komórek beta, pozwalając tym samym na wyrównanie stężenia glukozy wewnątrzkomórkowej i zewnątrzkomórkowej przez błonę komórkową. Metabolizm glukozy za pomocą glikolizy generuje ATP, a wyższy stosunek ATP/ADP powoduje zamknięcie zależnych od ATP kanałów potasowych (KATP), odpowiedzialnych za utrzymanie potencjału błonowego w spoczynku, uniemożliwiając jonowi potasu przejście przez błonę komórkową. Następujący wzrost dodatniego ładunku wewnątrzkomórkowego prowadzi do depolaryzacji błony komórkowej, co skutkuje otwarciem napięciowo-zależnych kanałów wapniowych i wzrostem stężenia wapnia wewnątrzkomórkowego [3]. Glukozowy sygnał Ca2+i jest zorganizowany w synchroniczny i jednolity wzór oscylacyjny [4], prowadząc do pulsacyjnego wydzielania insuliny [5]. Ostatecznym efektem jest eksport insuliny z komórek beta do krążenia ogólnego.
Układ endokannabinoidowy (ECS) obejmuje receptory kannabinoidowe sprzężone z białkiem G [receptor CB1 (CB1R) i receptor CB2 (CB2R)], ich endogenne ligandy lipidowe lub endokannabinoidy, takie jak arachidonoyletanolamid (AEA) czy anandamid oraz enzymy biosyntezujące i degradowane, jak szczegółowo opisano poniżej. Odkrycie ECS wywołało lawinę eksperymentalnych badań nad jego różnorodnymi funkcjami biologicznymi [6]. Ewidencja zarówno z badań przedklinicznych, jak i badań na ludziach wskazuje, że nadaktywność układu CB1R przyczynia się do rozwoju insulinooporności oraz obu typów cukrzycy, tj. typu 1 (T1D) i typu 2 (T2D) [7,8], a blokada CB1R wykazuje korzystne efekty w łagodzeniu otyłości/zespołu metabolicznego i jego powikłań, w tym T2D [9–11]. Oznacza to, że składniki ECS są obecne i funkcjonalne w tkankach istotnie zaangażowanych w kontrolę glikemii, w tym w trzustce endokrynnej, chociaż nie pojawiła się jeszcze zgoda co do lokalizacji komórkowej i dokładnej funkcji konkretnych składników ECS. W niniejszym artykule przedstawimy krótki przegląd obecnej wiedzy na temat roli ECS w funkcji i przeżyciu komórek beta, w związku z jego zaangażowaniem w T2D.
Najnowsze badania w dziedzinie biomedycyny wykazały integralną rolę układu endokannabinoidowego (ECS) w określaniu ryzyka sercowo-metabolicznego organizmu ludzkiego. Mechanizm ten jest pośredniczony poprzez wiązanie endokannabinoidów z receptorami CB1. Uważa się, że stymulacja receptorów CB1 w mózgu kontroluje i wpływa na apetyt. W normalnej fizjologii aktywacja receptorów CB1 odpowiada za homeostazę energetyczną, reguluje emocje oraz zachowania, takie jak lęk, strach, apetyt, spożycie jedzenia i wody. Receptory CB1 znajdują się również w tkankach obwodowych, takich jak wątroba, trzustka, mięśnie szkieletowe i tkanka tłuszczowa, odgrywające istotną rolę w metabolizmie lipidów i glukozy. Nadmierna aktywacja ECS wiąże się z różnymi chorobami metabolicznymi, takimi jak dyslipidemia, insulinooporność, lipogeneza, nadmierne przybieranie na wadze i zwiększenie otyłości brzusznej. Wszystkie te zdarzenia prowadzą do zwiększonego ryzyka sercowo-naczyniowego. Stosowanie selektywnego blokera receptora CB1, takiego jak rimonabant, wykazało redukcję obwodu talii, lepszą kontrolę glikemii, obniżenie poziomu triglicerydów, podniesienie poziomu cholesterolu HDL i ogólną redukcję całkowitej masy ciała. Lek ten został zalecany dla pacjentów z zespołem metabolicznym.
źródło: https://pubmed.ncbi.nlm.nih.gov/19385504/
Zarządzaj zgodami plików cookie
Używamy plików cookies w celu optymalizacji naszej witryny i naszych serwisów. Więcej informacji:
Funkcjonalne
Zawsze aktywne
Przechowywanie lub dostęp do danych technicznych jest ściśle konieczny do uzasadnionego celu umożliwienia korzystania z konkretnej usługi wyraźnie żądanej przez subskrybenta lub użytkownika, lub wyłącznie w celu przeprowadzenia transmisji komunikatu przez sieć łączności elektronicznej.
Preferencje
Przechowywanie lub dostęp techniczny jest niezbędny do uzasadnionego celu przechowywania preferencji, o które nie prosi subskrybent lub użytkownik.
Statystyka
Przechowywanie techniczne lub dostęp, który jest używany wyłącznie do celów statystycznych.Przechowywanie techniczne lub dostęp, który jest używany wyłącznie do anonimowych celów statystycznych. Bez wezwania do sądu, dobrowolnego podporządkowania się dostawcy usług internetowych lub dodatkowych zapisów od strony trzeciej, informacje przechowywane lub pobierane wyłącznie w tym celu zwykle nie mogą być wykorzystywane do identyfikacji użytkownika.
Marketing
Przechowywanie lub dostęp techniczny jest wymagany do tworzenia profili użytkowników w celu wysyłania reklam lub śledzenia użytkownika na stronie internetowej lub na kilku stronach internetowych w podobnych celach marketingowych.