
Nierównowaga jelitowa prowadzi do zapalnej choroby jelit (IBD), a przewlekły stan zapalny predysponuje do onkogenezy. Komórki dendrytyczne prezentujące antygen (DC) i makrofagi mogą przechylić równowagę w kierunku tolerancji lub patologii. Tutaj pokazujemy, że delta-9-tetrahydrokannabinol (THC) osłabia raka jelita grubego związanego z zapaleniem jelita grubego i zapalenie jelita grubego wywołane przez przeciwciała anty-CD40. Działając poprzez receptor kannabinoidowy 2 (CB2), THC zwiększa ekspresję CD103 na DC i makrofagach oraz zwiększa ekspresję TGF-β1, aby zwiększyć liczbę komórek regulatorowych T (Tregs). Komórki Tregs indukowane przez THC są niezbędne do zaradzenia systemicznemu IFNγ i TNFα wywołanemu przez przeciwciała anty-CD40, ale hamowanie APC przez CB2 przez THC gasi patogenne uwalnianie IL-22 i IL-17A w jelicie grubym. Badając tkanki z wielu miejsc, potwierdziliśmy, że THC wpływa na DC, szczególnie w miejscach bariery śluzowej w okrężnicy i płucach, aby zmniejszyć DC CD86. Używając modeli zapalenia okrężnicy i zapalenia układowego wykazujemy, że THC, poprzez CB2, jest silnym supresorem nieprawidłowych odpowiedzi immunologicznych, prowokując koordynację między APC i Treg.
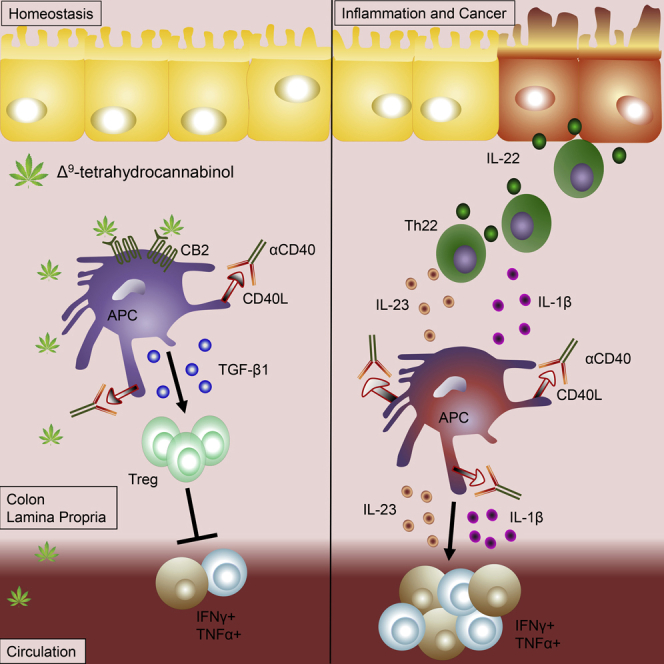
- THC może zapobiegać rozwojowi raka jelita grubego u myszy wywołanego zapaleniem okrężnicy
- •Aktywacja CB2 w komórkach układu odpornościowego tłumi chorobotwórczą onkogenną IL-22
- •THC osłabia zapalenie jelit wywołane przez anty-CD40 poprzez Tregs i APCs
- •Aktywacja CB2 na komórkach APC przez THC zmniejsza wydzielanie cytokin prozapalnych
Równowaga między tolerancją immunologiczną a uprzedzeniami w jelitach wymaga precyzyjnie dostrojonych sygnałów molekularnych, aby zapobiec wadom, które mogą prowadzić do chorób zapalnych jelit (IBD), choroby Leśniowskiego-Crohna i wrzodziejącego zapalenia jelita grubego ( Lynch i Pedersen, 2016 ). Stany te charakteryzują się nieograniczonym zapaleniem przewodu pokarmowego, które powoduje zniszczenie bariery śluzówkowej. Obciążenie IBD rośnie na całym świecie, a zapadalność i rozpowszechnienie zwiększają się szczególnie w krajach wschodnich i rozwijających się ( Coward i in., 2019 ). Wykazano jednoznacznie, że oprócz zgubnych skutków IBD osoby dotknięte tą chorobą mają zwiększone ryzyko zachorowania na raka jelita grubego (CRC) w ciągu swojego życia ( Dyson i Rutter, 2012 ). Chociaż pełna etiologia IBD jest niejasna, u podstaw jej rozwoju leży wyraźny składnik genetyczny ( Cho, 2008 ), a ostatnie badania dokładnie zmapowały 45 potencjalnych loci jako wyzwalające IBD ( Huang i in., 2017 ). Pomimo postępu w określeniu genetycznej podatności na IBD, czynniki genetyczne żywiciela nie mogą odpowiadać za rosnącą zapadalność na tę chorobę na całym świecie. Dlatego należy brać pod uwagę czynniki środowiskowe i mikrobiologiczne, a także odpowiedź immunologiczną na nie.
Główne geny podatności na IBD, w tym NOD2, STAT3 i IL-23 ( Cho, 2008 ; Huang i in., 2017 ; Duerr i in., 2006 ; de Souza i Fiocchi, 2016 ), są silnie zaangażowane w procesy immunologiczne. W związku z tym stężenia interleukiny (IL)-23, IL-17 i IL-22 występują na wyższych poziomach w surowicy i jelitach pacjentów z IBD ( Brand i in., 2006 ; Fujino i in., 2003 ). Prozapalne IL-17 i IL-23 odgrywają dobrze zdefiniowaną rolę w kaskadach zapalnych, które wywołują IBD; jednak rola IL-22 jest kwestią sporną. IL-22 jest kluczowa w regulacji powierzchni barierowych: zwiększaniu obrony nabłonkowej i śluzówkowej, uwalnianiu peptydów przeciwdrobnoustrojowych ( Parks i in., 2016 ) i ochronie komórek macierzystych enterocytów ( Gronke i in., 2019 ) i jest niezbędna do usunięcia zakażenia Citrobacter rodentium ( Manta i in., 2013 ). IL-22 została również zidentyfikowana jako patogenna ( Eken i in., 2014 ), a IL-22 jest silnym kandydatem do inicjowania CRC poprzez zwiększanie ekspresji STAT3 w komórkach nabłonkowych ( Sun i in., 2015 ; Kryczek i in., 2014 ; Kirchberger i in., 2013 ). Głównymi producentami IL-22 w jelitach są wrodzone komórki limfoidalne grupy 3 (ILC3) i komórki Th CD4+, które wydzielają IL-22 na niższych poziomach ( Dudakov i in., 2015 ). Produkcja IL-22 zarówno przez komórki ILC3, jak i Th22 może być stymulowana przez prozapalne cytokiny IL-23, MCP-1, IL-1β i IL-6 ( Parks i in., 2016 ; Manta i in., 2013 ; Dudakov i in., 2015 ). Produkcja IL-22 przez ILC3 jest ściśle kontrolowana przez czynniki egzogenne i endogenne. Metabolity z pożywienia i drobnoustrojów modulują czynniki transkrypcyjne, takie jak AhR, RORγt i STAT3 ( Kiss i Diefenbach, 2012 ). Endogenna aktywność komórkowa pomiędzy ILC3 a komórkami prezentującymi antygen (APC) może również regulować wydzielanie ILC3 IL-22. Komórki APC w jelitach są arbitrami pomiędzy tolerancją a opornością, przygotowując ILC, komórki Th i chroniące jelita komórki regulatorowe T (Tregs) przed uznanymi zagrożeniami. Komórki dendrytyczne (DC) są klasycznymi komórkami APC i prezentują antygeny komensalne i dietetyczne komórkom T w jelitach. Komórki DC w jelitach nie są jednorodne. Komórki DC CD103+ są zazwyczaj tolerogenne, wyrażając wysokie poziomy dehydrogenazy aldehydowej, TGF-β1 i integryny β8, które działają w celu indukowania komórek Tregs i regulacji w górę CCR9 w celu skierowania się do jelit ( Sun i in., 2007 ; Merad i in., 2013 ; Coombes i in., 2007). Uważa się jednak, że zwiększona ekspresja CD103 na DC odgrywa rolę w krzyżowym inicjowaniu wirusa i cytotoksycznych komórek T specyficznych dla nowotworów ( Hildner i in., 2008 ), a rola CD103+ CD11b+ DC jest mniej jasna, wykazując fenotyp miecza obosiecznego, w którym mogą zachowywać się jak CD103+ DC, wydzielając kwas retinowy i zwiększając liczbę Treg, lub mogą indukować komórki Th17 ( Persson i in., 2013 ). CD103- CD11b+ DC działają w bardziej klasyczny sposób prozapalny, podobnie jak makrofagi. Makrofagi w jelitach, zwłaszcza tkankowo rezydujące makrofagi CX 3 CR1+ (receptor fraktalkiny) (zwane również komórkami dendrytycznymi mieloidalnymi lub fagocytami jednojądrowymi) są dominującym źródłem cytokin prozapalnych, IL-23, IL-1β i czynnika stymulującego kolonie granulocytów i makrofagów (GM-CSF) w jelitach, które są silnymi aktywatorami produkcji ILC3 IL-22, a usunięcie tych komórek zmniejsza odsetek ILC3 wytwarzających IL-22 ( Manta i in., 2013 ; Longman i in., 2014 ).
Modulacja układu endokannabinoidowego (ECS) w przypadku IBD okazała się obiecująca w modelach przedklinicznych ( Whiting i in., 2015 ; Singh i in., 2012 ; Storr i in., 2009 ; Ke i in., 2016 ) i małych kohortach ludzkich ( Naftali i in., 2013 ; Lahat i in., 2012 ). Dane mysie dotyczące ECS w CRC wskazują na rolę kannabinoidów w zapobieganiu nieprawidłowemu tworzeniu krypt przez CB2 ( Izzo i in., 2008 ) i bezpośredniej apoptozie komórek nowotworowych przez CB1 lub CB2 ( Cianchi i in., 2008 ). Dane ludzkie wskazują, że CB2 jest nadregulowane w tkance guza jelita grubego, a guzy wyrażające wyższy poziom mRNA CB2 bardziej proliferują ( Martínez-Martínez i in., 2015 ). Rozbieżność między obserwacjami aktywności ECS w raku można częściowo wyjaśnić ustaleniami, że agoniści receptora kannabinoidowego (CB) wykazują bimodalne działanie w liniach komórek rakowych z odpowiedziami proliferacyjnymi przy niższych poziomach endogennych i odpowiedziami apoptotycznymi przy wyższych dawkach egzogennych ( Pisanti i in., 2013 ). W tym badaniu wykorzystujemy egzogenny kannabinoid delta-9-tetrahydrokannabinol (THC) w celu wpłynięcia na równowagę immunologiczną jelit w kierunku homeostazy w modelach stanu zapalnego nowotworowego i przednowotworowego. Wykazaliśmy, że THC jest skutecznym leczeniem w zapobieganiu rakowi jelita grubego związanemu z zapaleniem okrężnicy, wykorzystując model azoksymetanu (AOM) + siarczanu sodu dekstranu (DSS) w nowotworzeniu jelita grubego. Wykazaliśmy również, że pronowotworowa IL-22 w mikrośrodowisku nabłonka jest redukowana poprzez aktywację CB2 zależną od THC w komórkach hematopoetycznych. Aby wyizolować wpływ THC na układ odpornościowy i zbadać interakcje między odpornością adaptacyjną jelit a wrodzoną w celu regulacji wydzielania IL-22, zastosowaliśmy model anty-CD40, który inicjuje przebieg choroby niezależnie od mikrobioty lub przepuszczalności jelit w celu stymulacji stanu zapalnego ( Uhlig i in., 2006 ; Barthels i in., 2017 ). Interakcja CD40-CD154 (CD40L) między APC i limfocytami T jest ważna dla inicjacji i utrzymania adaptacyjnych odpowiedzi immunologicznych ( Cong i in., 2000 ). Blokowanie interakcji CD40-CD40L może być stosowane profilaktycznie i terapeutycznie w ustalonych modelach zapalenia jelita grubego u myszy ( Polese i in., 2002 ). U ludzi, komórki CD40+ APC znajdują się w pobliżu limfocytów T CD40L+ w zapalonej tkance IBD ( De Jong i in., 2000 ). Badania wykazały, że stymulacja CD40 u myszy inicjuje kaskadę zapalną zależną od produkcji IL-12 przez komórki mieloidalne w celu wywołania uwalniania interferonu (IFN)-γ przez limfocyty T i komórki NK, co powoduje stan zapalny układowy, oraz uwalnianie IL-23 przez mieloidalne prowadzące do zapalenia okrężnicy ( Uhlig i in., 2006). U myszy z nienaruszoną odpornością adaptacyjną, Tregs odgrywają ważną rolę w rozwoju i odporności na choroby. LAG-3+ Tregs mogą zmniejszyć kolitogenne stymulowane CD40 wydzielanie IL-23 przez makrofagi CX3CR1+, aby zapobiec patogennej produkcji ILC3 IL-22 ( Bauché i in., 2018 ). Odwrotnie, anty-CD40 działa na DC CD103+, aby zwiększyć regulację CCR7 i migrować do węzła chłonnego krezkowego (mLN), gdzie ulegają apoptozie, zmniejszając liczbę RORγt+ Foxp3+ Tregs, które prowadzą do śmiertelnego zapalenia jelita grubego wywołanego przez komórki T ( Barthels i in., 2017 ). W tym badaniu przyjrzano się niezliczonym mechanizmom wrodzonej i adaptacyjnej odporności w jelitach, aby dokładnie określić, w jaki sposób THC zapobiega rakowi jelita grubego związanemu z zapaleniem jelita grubego.
THC osłabia postęp raka jelita grubego wywołanego zapaleniem okrężnicy, który jest związany ze zmniejszeniem produkcji IL-22 w mikrośrodowisku nabłonka
Aby wywołać raka jelita grubego, zastosowaliśmy dobrze ugruntowany model wstrzyknięcia substancji rakotwórczej, AOM (10 mg/kg, dootrzewnowo [ip]), po którym nastąpiły trzy cykle DSS. Podawanie THC (10 mg/kg, doustnie przez sondę) lub nośnika (VEH, PBS:EtOH:Tween-80, w stosunku 17:2:1) rozpoczęto równocześnie z pierwszym cyklem DSS i podawano dwa razy w tygodniu. Leczenie THC i VEH przerwano po trzecim cyklu DSS, aby zbadać wpływ THC na inicjację raka, a nie potencjalne bezpośrednie skutki THC na guzy. Postęp choroby i schemat eksperymentalny są szczegółowo opisane w ( Rysunek 1A ), ujawniając, że myszy, którym podano DSS + AOM w celu wywołania raka jelita grubego (grupa CC), ale leczono THC, straciły mniej na wadze w porównaniu z grupą CC + VEH ( Rysunek 1A ). Po zakończeniu badania grupa CC + VEH wykazywała guzy jelita grubego, podczas gdy CC + THC nie wykazywało guzów ( Rysunek 1 B). Wielkość śledziony była również zwiększona w grupie CC + VEH, podczas gdy leczenie THC doprowadziło do znacznego zmniejszenia wielkości śledziony ( Rysunki 1 C i 1D). Kolonoskopie i barwienie H&E jelit wykazały zmniejszenie nasilenia stanu zapalnego i indukcji guza w grupie CC + THC w porównaniu z CC + VEH ( Rysunki 1 E–1G). Ponieważ przewlekła regulacja w górę IL-22 prowadzi do rozwoju raka, a u myszy CC + THC nie rozwinęły się żadne guzy, przyjrzeliśmy się parametrom odpornościowym, w szczególności IL-22. IL-22 to cytokina produkowana głównie przez komórki Th22 i ILC w jelicie, która ma kluczowe znaczenie dla rozwoju, utrzymania i macierzystości raka jelita grubego wywołanego stanem zapalnym ( Sun i in., 2015 ; Kryczek i in., 2014 ; Kirchberger i in., 2013 ). Aby scharakteryzować populacje komórek odpornościowych, komórki blaszki właściwej okrężnicy (cLP) i komórki śródnabłonkowe (IEC) poddano analizie cytometrii przepływowej. Strategie bramkowania są szczegółowo opisane na rysunkach S1 A–S1D. Grupa CC + VEH miała znaczący wzrost liczby komórek Th22 CD4+IL-22+ w IEC okrężnicy w porównaniu z CC + THC, podczas gdy nie było zmiany w komórkach cLP CD4+ RORγt+ ( Rysunki 1 H–1J). Pobranie komórek z cLP i IEC i umieszczenie ich na noc w celu zebrania supernatantów wykazało, że wzrost IL-22 obserwowany w grupie CC + VEH w mikrośrodowisku okrężnicy pochodził konkretnie z komórek w IEC ( rysunki 1 K i 1L), a nie z cLP. Powyższe dane wykazały, że THC zapobiega rozwojowi raka jelita grubego w modelu DSS + AOM i że jest to związane ze spadkiem liczby IEC produkujących IL-22.
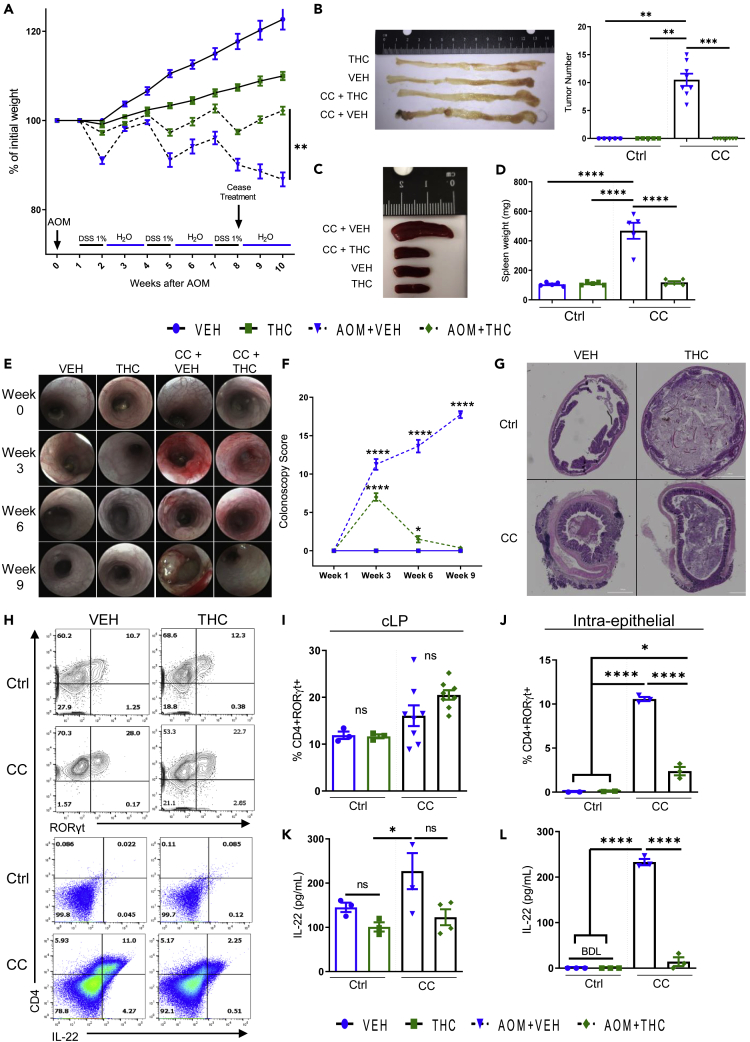
Aktywacja receptora kannabinoidowego hamuje postęp raka jelita grubego wywołanego zapaleniem okrężnicy poprzez zmniejszenie produkcji IL-22 w mikrośrodowisku nabłonka
Aby wywołać raka jelita grubego związanego z zapaleniem jelita grubego (CC), myszom podano pojedynczą dawkę AOM, ip (10 mg/kg), a następnie tydzień później rozpoczęto leczenie równocześnie z indukcją pierwszego cyklu zapalenia jelita grubego z 2% DSS w wodzie pitnej. Tygodniowe cykle DSS (2%) były kontynuowane przez 2 tygodnie regularnego picia wody przez 3 cykle trwające 9 tygodni. Leczenie VEH (10% EtOH w PBS-Tween-80), THC (10 mg/kg) lub kombinacją THC i CBD (10 mg/kg, oba) było podawane dwa razy w tygodniu aż do zakończenia ostatniego cyklu DSS, a następnie leczenie zostało przerwane w celu monitorowania wpływu kannabinoidów na indukcję raka, a nie bezpośredniego wpływu kannabinoidów na sam nowotwór. Myszy kontrolne były leczone dwa razy w tygodniu, ale choroba nie została wywołana (ctrl).
(A) Schemat przedstawiający procentową zmianę masy ciała, schemat leczenia i przebieg choroby. (n = 5, grupy kontrolne; n = 7–9 grup CC).
(B) Reprezentatywne zdjęcie i liczba guzów w każdym okrężnicy w momencie poświęcenia. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i są przedstawione jako średnia ± SEM; ∗∗p < 0,01, ∗∗∗p < 0,001 za pomocą testu Kruskala-Wallisa z testem wielokrotnych porównań Dunna.
(C i D) (C) Fotografia reprezentatywna i (D) ilościowe określenie masy śledziony u wskazanych myszy w momencie uśmiercenia.
(E i F) Kolonoskopie wykonywano przez cały okres trwania eksperymentu, a obrazy reprezentatywne przedstawiono w (E) i ilościowo w (F) (n = 5–8, grupy kontrolne; n = 8 grupy CC).
(G) Typowe obrazy H&E okrężnic w momencie uśmiercenia. cLP i frakcję komórek śródnabłonkowych (IEC) wyizolowano w momencie uśmiercenia i barwiono w celu wykrycia komórek CD4+ RORγt+ i CD4+ IL-22+.
(H) Typowe wykresy konturowe przedstawiające komórki Th17 CD4+ RORγt+ (bramka: żywe, CD45+ CD3+ CD4+) w cLP (dwa górne panele) i komórki Th22 CD4+ IL-22+ (bramka: żywe, CD45+ CD3+) we frakcji IEC (dwa dolne panele).
(I i J) Kwantyfikacja procentów z wykresów cytometrii przepływowej w (H), uzyskanych z (I) clP i (J) IEC (n = 3, grupy kontrolne; n = 3–8, grupy CC).
Komórki (K i L) 1 × 106 pochodzące z warstw (K) cLP lub (L) IEC ze wskazanych grup umieszczono na płytkach na noc, zebrano supernatanty i poddano testom ELISA na obecność IL-22 (n = 3–4).
Każdy symbol reprezentuje pojedynczą mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i są przedstawione jako średnia ± SEM NS, nieistotne: ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,001, ∗∗∗∗p < 0,0001 za pomocą dwukierunkowej analizy wariancji z testem wielokrotnych porównań Tukeya.
Komórki hematopoetyczne i niehematopoetyczne przyczyniają się do ochrony przed zapaleniem jelita grubego wywołanym przez THC, ale komórki hematopoetyczne są niezbędne do zmniejszenia produkcji IL-22
Aby ustalić, czy ekspresja CB na komórkach odpornościowych czy komórkach niehematopoetycznych była odpowiedzialna za korzystne efekty THC i określić mechanizm, poprzez który THC może zmniejszyć wydzielanie nowotworowe IEC IL-22, myszy dzikiego typu (WT) poddano mieloablacji i zrekonstruowano szpikiem kostnym (BM) myszy CB2KO (CB2KO→WT) lub WT BM (WT→WT), ponieważ zdecydowana większość ekspresji CB na komórkach odpornościowych to CB2. Następnie wywołaliśmy ostre zapalenie jelita grubego za pomocą wstrzyknięcia AOM i pojedynczego cyklu DSS, aby modelować wczesne stadia tumorigenezy przed makroskopowym rozwojem guza, ale gdzie występuje charakterystyczna utrata masy ciała, skrócenie jelita grubego i stan zapalny. W obu grupach, które otrzymywały THC, utrata masy ciała i skrócenie jelita grubego wywołane przez DSS zostały zniesione w porównaniu z kontrolami VEH, a skrócenie jelita grubego było bardziej wyraźne w grupie CB2KO → WT ( Rysunki 2 A i 2B). Co ciekawe, myszy CB2KO→WT, którym podano THC, miały zmniejszoną liczbę komórek Th22 w cLP w porównaniu z myszami VEH i WT→WT, ale tylko myszy WT→WT, którym podano THC, miały zmniejszoną liczbę komórek IEC Th22 ( rysunki 2 C i 2D). Wydzielanie IL-22 przez cLP ILC3 nie uległo zmianie pod koniec eksperymentu ( rysunek S1 E). Wpływ THC nie miał bezpośredniego wpływu na komórki Th22, ponieważ polaryzacja Th22 komórek naiwnych w obecności THC nie wpływała na wydzielanie IL-22 ( rysunek 2 E). Ponieważ wykazaliśmy, że THC może znieść indukcję aberracyjnej IL-22 w ostrych i przewlekłych modelach tumorigenezy i że wpływ ten jest niezależny od bezpośredniego wpływu THC na generację Th22, przeszliśmy do modelu, w którym stymulacja produkcji IL-22 jest pośredniczona przez produkcję prozapalnych cytokin przez komórki mieloidalne.
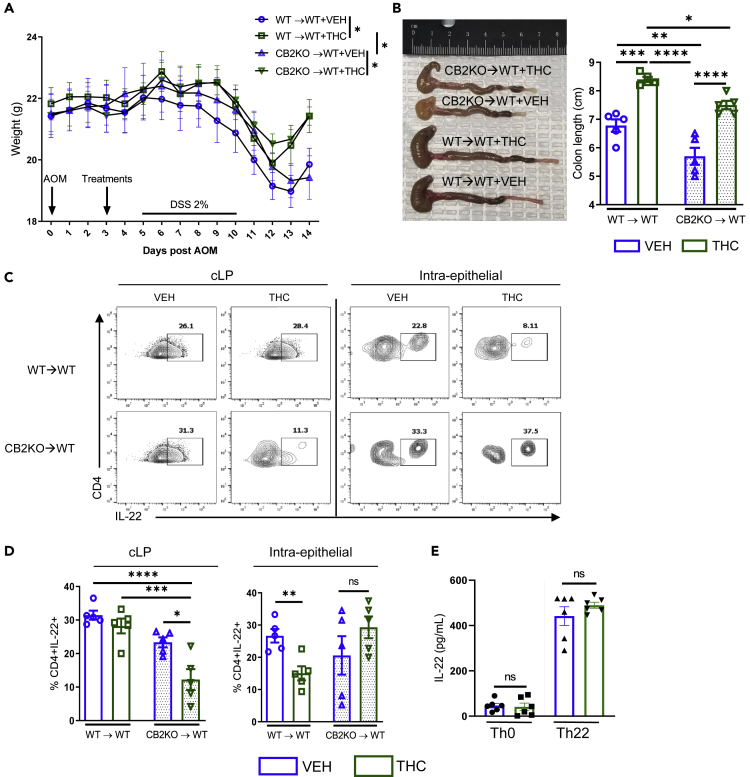
Komórki hematopoetyczne i niehematopoetyczne przyczyniają się do ochrony przed zapaleniem jelita grubego wywołanym przez THC, ale komórki hematopoetyczne są niezbędne do zmniejszenia produkcji IL-22
U myszy WT przeprowadzono ablację mieloblasty za pomocą dwóch dawek po 600 cGy w odstępie 3 godzin, a następnie przeprowadzono odbudowę układu odpornościowego poprzez przeniesienie komórek szpiku kostnego od myszy WT (WT → WT) lub myszy Cnr2−/−, z wyłączonym genem CB2 (CB2 → WT).
(A) Masa ciała w trakcie trwania choroby (n = 4–5).
(B) Długości okrężnicy na zakończenie eksperymentu.
(C i D) (C) Typowe wykresy konturowe przedstawiające komórki Th22 CD4+ IL-22+ (bramka: Żywe, CD45+ CD3+) we frakcji cLP IEC. (D) Ilościowe oznaczenie komórek ekspresujących CD4+ IL-22 w blaszce właściwej jelita grubego i frakcjach nabłonkowych (n = 4–5).
(E) Naiwne komórki T CD4+ wyizolowano od poszczególnych myszy i umieszczono w warunkach polaryzacji Th0 lub Th22 z VEH lub THC (10 μM) przez 3 dni. Zebrano supernatanty i poddano je testowi ELISA na obecność IL-22 (n = 6).
Każdy symbol reprezentuje pojedynczą mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i są przedstawione jako średnia ± SEM; NS, nieistotne statystycznie; ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,001, ∗∗∗∗p < 0,0001 metodą dwukierunkowej analizy wariancji z testem wielokrotnych porównań Tukeya.
Leczenie THC zmniejsza nasilenie zapalenia okrężnicy i całego układu wywołanego przez αCD40
Aby zbadać mechanizm, poprzez który THC zmniejsza produkcję IL-22 w jelitach, przeszliśmy do modelu zapalenia jelit przeciwko CD40 (αCD40). Wstrzyknięcie αCD40 powoduje ostre zapalenie jelit zależne od produkcji IL-22 ( Eken i in., 2014 ) i ogólnoustrojowy stan zapalny, który objawia się ostrą wyniszczającą chorobą i hiperaktywacją komórek zapalnych. Podawanie THC (10 mg/kg, doustnie przez sondę) rozpoczynające się 3 dni przed indukcją choroby nie mogło zmniejszyć utraty masy ciała związanej z początkiem choroby ( Rysunek S1 F). THC zmniejszyło splenomegalię występującą 7 dni po wstrzyknięciu αCD40 ( Rysunek 3 A) i zmniejszyło krążące prozapalne cytokiny IFNγ, IL-17A, IL-22 i TNFα w szczytowym momencie choroby 3 dnia; cytokiny związane z Th2 pozostały niezmienione ( Rysunki 3 B i 3C). Histologicznie THC zmniejszyło naciek komórek zapalnych i uszkodzenia spowodowane przez αCD40 w jelicie grubym i wątrobie ( rycina 3 D). Efekt ten był wyraźnie widoczny w jelicie grubym za pomocą kolonoskopii ( ryciny 3 E i 3F); uszkodzenie wątroby wywołane przez αCD40 mierzono, badając aktywność enzymatyczną aminotransferazy asparaginianowej (AST) i aminotransferazy alaninowej (ALT) w surowicy. Myszy αCD40, którym podano kontrole VEH, miały wyższą aktywność AST trzeciego dnia niż kontrole IgG, a myszy αCD40, którym podano THC, nie wykazywały tego wzrostu; jednak zarówno myszy leczone VEH, jak i THC miały wyższą aktywność ALT trzeciego dnia niż kontrole IgG ( ryciny 3 G i 3H).
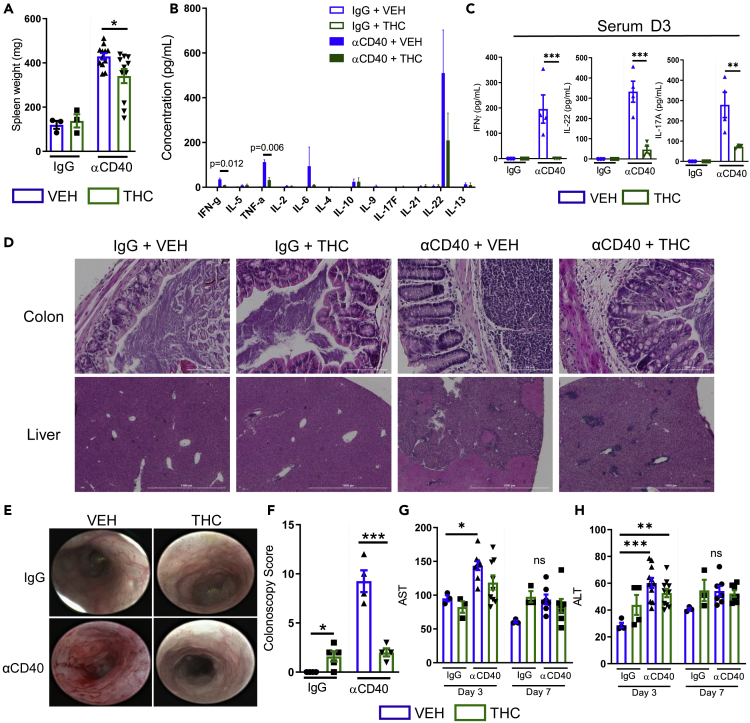
Leczenie THC zmniejsza nasilenie zapalenia okrężnicy i całego układu wywołanego przez αCD40
Myszom podawano codziennie VEH lub THC (10 mg/kg) przez 3 dni przed dootrzewnowym wstrzyknięciem szczurzego przeciwciała anty-mysiego IgG (kontrola) lub szczurzego przeciwciała anty-mysiego αCD40 (200 μg, klon FGK4.5 w PBS), a leczenie kontynuowano przez 7 dni po wywołaniu choroby w celu monitorowania postępu ciężkości stanu zapalnego.
(A) Masa śledziony (n = 3–10).
(B) Trzeciego dnia pobrano krew poprzez krwawienie z oczodołu, oddzielono surowicę i poddano ją badaniu Legendplex w celu określenia poziomu cytokin pomocniczych T w surowicy (n = 3 w każdej grupie).
(C) Testy ELISA dla IFNγ, IL-22 i IL-17A z surowicy pobranej trzeciego dnia od myszy, którym podano leczenie lub VEH 3 dni przed wywołaniem choroby.
(D) Przykładowe obrazy H&E jelita grubego (górny rząd, powiększony 20X) i wątroby (dolny rząd, powiększony 4X).
(E i F) (E) Obrazy reprezentatywne z kolonoskopii wykonanych trzeciego dnia i ich ocena ilościowa (F) (n = 4 w każdej grupie).
(G i H) Ilościowe oznaczenie aktywności (G) AST i (H) ALT mierzonej w surowicy 3. i 7. dnia po wstrzyknięciu αCD40.
Każdy symbol reprezentuje pojedynczą mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla czterech niezależnych eksperymentów i są przedstawione jako średnia ± SEM. NS, nieistotne statystycznie; ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,005 w dwukierunkowej analizie wariancji z testem wielokrotnych porównań Tukeya.
Leczenie THC zmniejsza naciek komórek zapalnych jelita grubego i zwiększa liczbę Tregów Lamina Propria
Następnie staraliśmy się zbadać mechanizmy komórkowe w okrężnicy, poprzez które działa THC, aby zmniejszyć stan zapalny okrężnicy wywołany przez αCD40. Leczenie THC u myszy αCD40 skutkowało zmniejszeniem limfadenopatii krezkowej i zmniejszeniem odsetka komórek odpornościowych CD45+ migrujących do cLP, chociaż THC nie zmniejszyło całkowitej liczby komórek odpornościowych ( rysunki 4 A i 4B). Wstrzyknięcie αCD40 skutkowało ostrym uwolnieniem IL-22, które wbrew intuicji ochronnej roli IL-22 w modelach infekcji i chemicznych ( Gronke i in., 2019 ; Manta i in., 2013 ) jest patogenne na tych poziomach ( Eken i in., 2014 ). THC zmniejszyło wydzielanie IL-22 z komórek cLP u myszy αCD40 ( rysunek 4 C). Biorąc pod uwagę, że badanie aktywności cytokin z surowicy w dniu 3 ( Rysunek 3 B) nie wykazało wzrostu cytokin przeciwzapalnych IL-4, IL-10 lub IL-9 po leczeniu THC, rozważyliśmy mechanizmy supresji αCD40 niezależne od cytokin Th2. Zbadaliśmy komórki APC cLP i odkryliśmy, że THC zmniejszyło liczbę makrofagów cLP CD11b+ F4/80+, ale nie zmieniło liczby komórek DC cLP MHCII HI CD11c+ (strategie bramkowania opisano szczegółowo na rysunkach S1 A–S1D) ( Rysunki 4 D i 4E). Pomimo zmniejszenia liczby makrofagów cLP obserwowanego po leczeniu THC, markery aktywacji CD80 i CD86 nie uległy zmianie u myszy αCD40 otrzymujących THC, ale ekspresja komórek DC CD80 została zmniejszona po podaniu THC ( Rysunek S2 A–S2D). Aby ocenić, jaką rolę w chorobie odgrywają komórki DC cLP, scharakteryzowaliśmy podzbiory komórek DC, o których wiadomo, że silnie wpływają na równowagę zapalną jelit. Fenotypowaliśmy DC pod kątem ekspresji markerów powierzchniowych CD103 i CD11b, ponieważ wykazano, że DC z wyższą ekspresją CD103 odgrywają rolę w promowaniu odpowiedzi przeciwzapalnej poprzez indukcję Treg ( Coombes i in., 2007 ), podczas gdy DC z wyższą ekspresją CD11b są katalizatorami odpowiedzi zapalnych komórek T i ILC ( Persson i in., 2013 ). Leczenie THC spowodowało wzrost liczby CD103+ DC i równoczesny spadek CD103+ CD11b + DC w momencie uśmiercenia w porównaniu z myszami VEH ( Rysunek 4 F). Naturalne FoxP3+Helios + Treg wzrosły z αCD40 w porównaniu z IgG, a THC dodane do tego efektu w porównaniu z VEH ( Rysunek 4 G). W przypadku mLN leczenie THC spowodowało zmniejszenie liczby komórek CD8+ CTL wydzielających IFNγ i komórek CD4+ Th1 w porównaniu z VEH ( rysunki S2 E–S2H), natomiast nie zaobserwowano różnic w komórkach CD4+ wydzielających IL-17A, IL-10 lub IL-4 ani w fenotypie komórek DC ( rysunki S2 G–S2L).
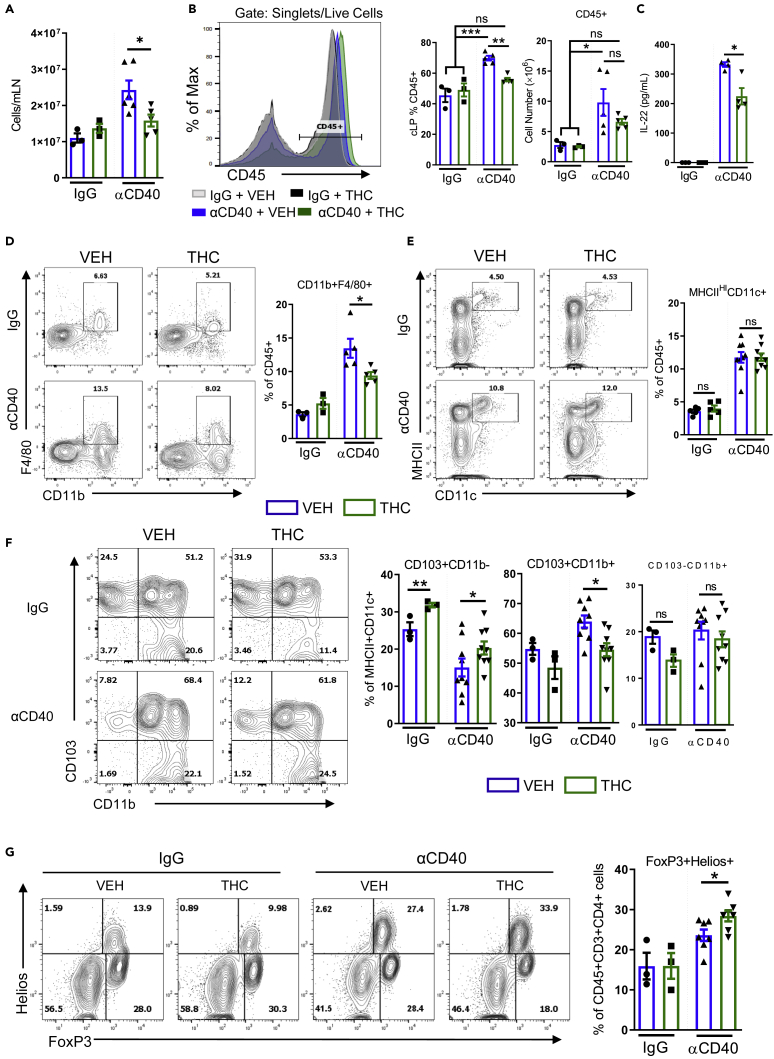
Leczenie THC zmniejsza naciek komórek zapalnych jelita grubego i zwiększa liczbę Tregów Lamina Propria
(A) Bezwzględna liczba komórek węzłów chłonnych krezkowych (n = 3–6).
(B) Typowe histogramy nałożone na cytometrię przepływową, wartości procentowe i bezwzględna liczba komórek CD45+ z blaszki właściwej jelita grubego wskazanych myszy.
(C) Wyniki testu ELISA dla IL-22 z supernatantów cLP pobranych od wskazanych myszy w dniu 7 (n = 3–4).
(D–F) (D) Typowe wykresy konturowe cytometrii przepływowej makrofagów (bramka: żywe, CD45+) (n = 3–5), (E) komórek dendrytycznych (bramka: żywe, CD45+) (n = 5–8) i (F) podzbiorów komórek dendrytycznych cLP w 7. dniu (bramka: żywe, CD45+ MHCII HI CD11c+) (n = 3–9).
(G) Typowe wykresy konturowe cytometrii przepływowej dla n- i iTreg w cLP (bramka: Żywe, CD45+ CD3+ CD4+) (n = 3–8).
Każdy symbol reprezentuje pojedynczą mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla czterech niezależnych eksperymentów i są przedstawione jako średnia ± SEM. ns, nieistotne: ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,005 według dwukierunkowej analizy wariancji z testem wielokrotnych porównań Tukeya.
Aktywacja CB2 w komórkach dendrytycznych zmniejsza markery aktywacji i zwiększa produkcję TGF-β1
Zmianę fenotypu DC wywołaną przez THC obserwowano również w chemicznych modelach zapalenia okrężnicy wywołanego roztworem kwasu pikrylosulfonowego (TNBS) i DSS ( rysunki S3 A i S3B), zatem aby zobaczyć rolę DC w ochronie przed zapaleniem okrężnicy wywołanym przez THC, użyto naiwnych myszy. Myszom WT podano pojedynczą dawkę (1X) VEH lub THC, a 24 godziny później przeanalizowano cLP DC, ujawniając, że THC zwiększyło ekspresję DC CD103 i zmniejszyło ekspresję CD11b w warunkach podstawowych ( rysunek 5 A). Używając myszy WT, Cnr1 −/− i Cnr2 −/− , THC zwiększyło ekspresję CD103 u wszystkich genotypów myszy; jednak naiwne Cnr2 −/− miały obniżone ogólne poziomy cLP DC w porównaniu z myszami WT i Cnr1 −/− ( rysunek S4 A). Zgodnie z tym zauważyliśmy zależny od CB2 wzrost FoxP3+ Helios+ nTregs po podaniu THC ( Rysunek 5 B). Zwiększenie ekspresji receptora chemokiny 7 (CCR7) jest ważne dla transportu DC między tkankami limfatycznymi ( Schulz i in., 2009 ) i zostało zgłoszone jako jeden z mechanizmów, poprzez który DC mogą inicjować Treg w cLP. DC w mLN i cLP myszy, którym podano 1X THC, miały zmniejszoną ekspresję CCR7 w porównaniu z kontrolami VEH ( Rysunek 5 C). Innym mechanizmem, poprzez który CD103+ DC mogą wpływać na indukcję Treg, jest wydzielanie TGF-β1 ( Coombes i in., 2007 ), a supernatanty odzyskane z komórek cLP zebranych od myszy THC lub VEH 1X i wysianych na noc wykazały zwiększoną całkowitą produkcję TGF-β1 u myszy leczonych THC ( Rysunek 5 D). Nie zauważono wzrostu liczby komórek DC CD103+ w cLP po ostrym podaniu THC w mLN tych myszy ( Rysunek S4 B), ani nie zauważono wzrostu całkowitej produkcji TGF-β1 w mLN ( Rysunek S4 C). Aby wyizolować efekty THC konkretnie na komórki DC, wygenerowano komórki dendrytyczne szpiku kostnego (BMDC) przez dodanie GM-CSF i IL-4 do hodowli naiwnych komórek BM. VEH lub THC dodano in vitro w momencie polaryzacji, a za każdym razem zmieniano medium, aby ujawnić, że pod koniec hodowli komórki BMDC traktowane THC miały więcej całkowitego TGF-β1 w swoim supernatancie ( Rysunek 5 E). Chociaż w hodowlach traktowanych VEH i THC występowały równoważne procenty DC, hodowla BMDC jest mieszaną populacją komórek, więc 6. dnia hodowli BMDC oczyszczono DC CD11c+ i umieszczono je na noc bez dodatkowego VEH lub THC, odzyskano ich supernatant, a całkowite poziomy TGF-β1 zostały przeanalizowane i wzrosły w przypadku BMDC traktowanych THC w porównaniu z kontrolami VEH ( Rysunek 5 E). Zaobserwowaliśmy spadek ekspresji DC CD86, ale nie CD80 w hodowlach traktowanych THC; DC traktowane THC zmniejszyły proliferację komórek T CD4 i CD8 in vitro (Ryciny S4 D i S4E). Potwierdziliśmy te wyniki u myszy naiwnych poprzez sekwencjonowanie RNA pojedynczych komórek (scRNA-seq) komórek okrężnicy 24 godziny po podaniu VEH lub THC ( rycina S3 C). scRNA-seq pozwoliło na wyizolowanie komórek mieloidalnych z innych typów komórek, ujawniając, że THC zmniejsza ekspresję Cd80 i Cd86 w komórkach mieloidalnych , ale zwiększa ekspresję Cd40 ( rycina 5 F).
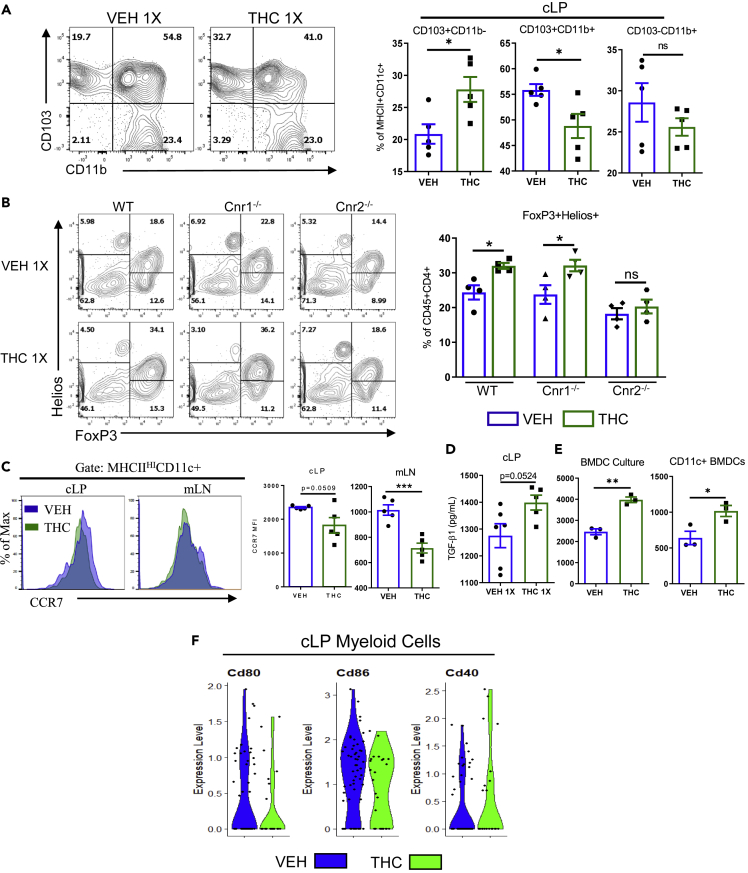
Aktywacja CB2 w komórkach dendrytycznych zmniejsza markery aktywacji i zwiększa produkcję TGF-β1
Naiwnym myszom podano VEH lub THC (10 mg/kg) jednorazowo, a po 24 godzinach pobrano cLP i mLN.
(A) Typowe wykresy konturowe cytometrii przepływowej podzbiorów komórek dendrytycznych w cLP.
(B) Wykresy konturowe cytometrii przepływowej przedstawiające FoxP3+ Helios+ nTregs i FoxP3+ Helios- iTregs (bramka: Live, CD45+ CD4+) (n = 4).
(C) Typowe histogramy nałożone na cytometrię przepływową oraz mediana intensywności fluorescencji (MFI) ekspresji CCR7 w komórkach DC u wskazanych myszy w cLP lub mLN (n = 5).
(D i E) (D) Całkowite poziomy TGF-β1 z supernatantów komórek cLP pochodzących od myszy leczonych VEH lub THC przez 24 godziny (n = 5–6) i (E) BMDC leczone VEH lub THC po 7 dniach hodowli z leczeniem lub po 1 dniu hodowli po selekcji komórek CD11c+ (n = 3). Każdy symbol reprezentuje pojedynczą mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i przedstawione są jako średnia ± SEM. NS, nieistotne statystycznie: ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,005 metodą dwukierunkowej analizy wariancji z testem wielokrotnych porównań Tukeya.
(F) Wykres skrzypcowy scRNA-seq obrazujący ekspresję mRNA w klastrze komórek mieloidalnych z okrężnicy myszy leczonych VEH lub THC przez 24 godziny.
THC indukuje Tregs poprzez DC, aby zmniejszyć stan zapalny ogólnoustrojowy, ale nie jelitowy
Aby rozszyfrować, czy THC działało bezpośrednio na limfocyty T, czy poprzez DC, stymulując Treg, limfocyty T CD4+ wyizolowano od myszy naiwnych i umieszczono je w warunkach stymulacji limfocytów T (αCD3 i αCD28), polaryzacji Treg (αCD3, αCD28, IL-2 i TGF-β1) lub z komórkami APC CD11c+, z THC lub bez. Tylko wtedy, gdy limfocyty T były umieszczone na płytkach z komórkami APC CD11c+, THC zwiększyło odsetek Treg ( Rysunek 6 A); jednak THC działało bezpośrednio na spolaryzowane Treg, zwiększając ekspresję powierzchniową markerów funkcjonalnych TGF-β1 (LAP) i CD25 ( Rysunek 6 B), ale GARP, ICOS i CD223 (LAG-3) pozostały niezmienione ( Rysunek S4 F).
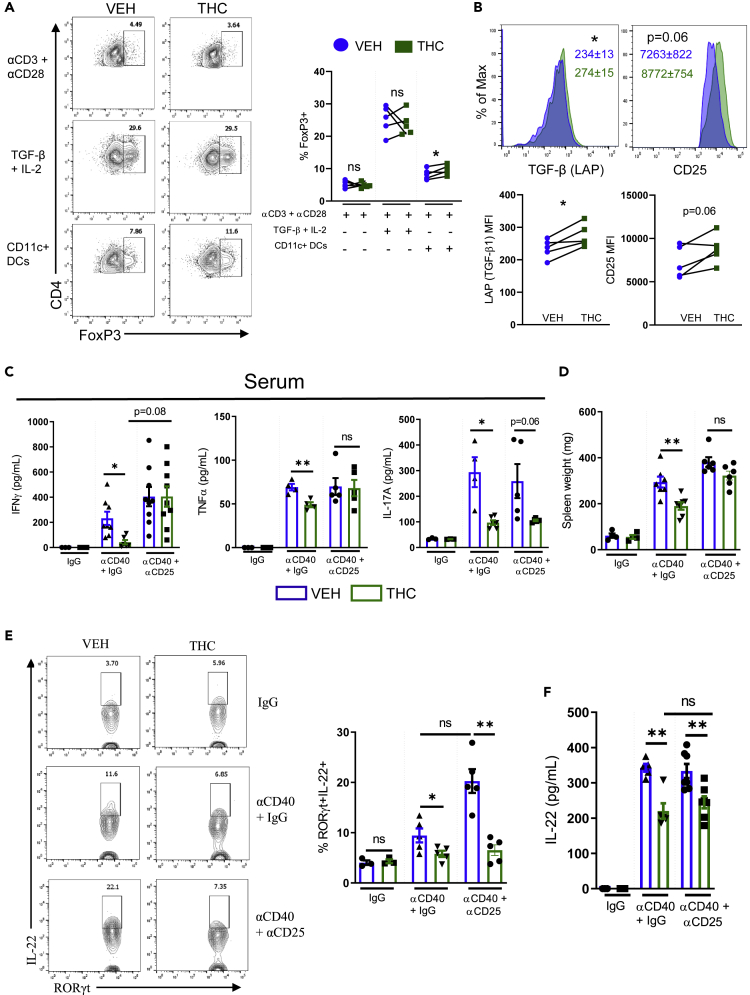
THC indukuje Tregs poprzez DC, aby zmniejszyć stan zapalny ogólnoustrojowy, ale nie jelitowy
Węzły chłonne i śledziony pobrano od myszy naiwnych, a komórki T CD4+ i APC CD11c+ wybrano poprzez oczyszczanie za pomocą kulek magnetycznych. Oczyszczone komórki T CD4+ umieszczono na płytkach na 5 dni w warunkach stymulacji komórek T: (αCD3 + αCD28), polaryzacji Treg: (αCD3 + αCD28 + IL-2 + TGF-β1) lub z oczyszczonymi komórkami CD11c+ w stosunku 5:1, CD4+:CD11c+, a następnie próbki od poszczególnych myszy potraktowano VEH lub THC (10 μM).
(A) Wykresy konturowe cytometrii przepływowej i ilościowe oznaczenie limfocytów Treg CD4+ FoxP3+ (bramka: żywe, CD4+) po 5 dniach hodowli w określonych warunkach (n = 5).
(B) Reprezentatywne nałożone histogramy wskazanych markerów z komórek CD4+ T leczonych THC lub VEH po polaryzacji Treg (bramka: Żywe, CD4+ FoxP3+) (n = 5). Każdy symbol reprezentuje sparowane porównanie z pojedynczej myszy, której komórki podzielono na leczone VEH lub THC. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i przedstawiono jako średnia ± SEM. NS, nieistotne: ∗p < 0,05, ∗∗p < 0,01 w teście t-Studenta. 10 dni przed indukcją choroby Treg zostały wyczerpane poprzez wstrzyknięcie dootrzewnowe szczurzego przeciwciała anty-mysiego CD25 (klon PC61, 250 μg/mysz) lub kontroli izotypowej. Myszy były wstępnie leczone VEH lub THC (10 mg/kg) przez 2 dni przed indukcją choroby poprzez wstrzyknięcie dootrzewnowe αCD40 lub kontroli IgG.
(C) ELISA na obecność cytokin w surowicy IFNγ, TNFα i IL-17A (n = 3–6).
(D) Masa śledziony na koniec eksperymentu (n = 3–6).
(E) Typowe wykresy konturowe cytometrii przepływowej i ilościowe oznaczenie wydzielających IL-22 komórek ILC3 (bramka: Żywa, linia-CD45 INT CD90.2 HI CD3- RORγt+) (n = 3–6).
(F) Wyniki testu ELISA dla IL-22 z supernatantów cLP pobranych od wskazanych myszy w dniu 7 (n = 3–5).
Każdy symbol reprezentuje pojedynczą mysz. Dane przedstawiono jako średnią ± SEM z jednego eksperymentu, który powtórzono 3 razy. NS, nieistotne: ∗p < 0,05, ∗∗p < 0,01 w dwukierunkowej analizie wariancji z testem wielokrotnych porównań Tukeya.
Aby ocenić rolę, jaką Treg odgrywają w osłabianiu procesów zapalnych αCD40 przez THC, Treg usunięto u myszy poprzez dootrzewnową iniekcję przeciwciała anty-CD25, które, jak wykazano, zmniejsza liczbę Treg 8 dni po pojedynczej iniekcji ( Setiady i in., 2010 ). Nie występowały one w mLN myszy leczonych przeciwciałami anty-CD25, ale nie u myszy kontrolnych IgG lub leczonych przeciwciałami αCD40+ IgG ( Rysunek S5 B). Leczenie THC lub VEH rozpoczęto 5 dni po podaniu przeciwciał anty-CD25 lub IgG i 2 dni przed αCD40. Ani myszy pozbawione Treg, którym podano αCD40, ani nie wykazywały większej utraty masy ciała w porównaniu z myszami αCD40+ IgG, ani THC nie zmieniało wyniszczenia ( Rysunek S5 A). THC zmniejszyło poziom IFNγ i TNFα w surowicy tylko u myszy αCD40+ IgG, nie u myszy αCD40 pozbawionych Treg; zmniejszenie poziomu Treg nie zmieniło zdolności THC do zmniejszania poziomu IL-17A w surowicy indukowanego przez αCD40 ( Rysunek 6 C). Utrata Treg zapobiegła zmniejszeniu masy śledziony obserwowanemu w przypadku THC+ αCD40 IgG ( Rysunek 6 D). Kolitogenna produkcja ILC3 IL-22 w modelu αCD40 jest częściowo regulowana przez Treg ( Bauché i in., 2018 ); jednak nasze wyniki wskazują, że THC zmniejsza wydzielające IL-22 ILC3, a także całkowitą produkcję IL-22 w okrężnicy niezależnie od Treg ( Rysunki 6 E i 6F).
THC zmniejsza markery aktywacji APC w jelicie grubym poprzez receptory kannabinoidowe
Ponieważ pokazaliśmy, że Tregs indukowane przez THC nie są pośrednikiem zapobiegającym zapaleniu okrężnicy, staraliśmy się przeanalizować inne zmiany komórek odpornościowych, które zachodzą po ostrym podaniu THC w warunkach naiwnych. Odkryliśmy, że THC nie wpływa na procent ILC: ILC2, NCR lub LTi ILC3 w cLP, ani nie zmienia się liczba makrofagów, chociaż występuje wzrost komórek tucznych FCεRI+ c-Kit+ w cLP po podaniu THC, a jest to pośredniczone przez CB2 ( Rysunki S5 C i S5D). Myszy leczone THC1X, których cLP wyizolowano i umieszczono na płytkach przez noc, wykazują jedynie niewielkie różnice w zakresie wzrostu produkcji IL-2 i IL-6 w porównaniu z myszami leczonymi VEH ( Rysunek S5 E). Biorąc pod uwagę globalną redukcję cytokin prozapalnych obserwowaną w przypadku THC po podaniu αCD40, przyjrzeliśmy się komórkom mieloidalnym w różnych tkankach, aby sprawdzić, w jaki sposób THC wpływa na ich immunogenność. cLP, mLN, śledziona, płuca, wątroba i mózg naiwnych myszy WT, Cnr1 −/− i Cnr2 −/− zostały pobrane, doprowadzone do zawiesiny pojedynczych komórek i umieszczone na noc w obecności VEH lub THC (10 μM), a następnie przeanalizowane cytometrią przepływową pod kątem interesujących markerów aktywacji APC: CD40, CD80, CD86, Ly-6C, Ly-6G, CD103, CX3CR1 i MHCII. Mapy cieplne przedstawiają medianę intensywności fluorescencji (MFI) dla każdego interesującego markera bramkowanego na makrofagach (Macs) lub DC z tej tkanki (strategie bramkowania dla poszczególnych tkanek przedstawiono na rysunkach S1 A–S1D) ( rysunek 7 A). Mapy cieplne są normalizowane dla każdego rzędu tak, aby zwiększone MFI silnie ekspresjonowanych markerów (np. MHCII na DC) nie przyćmiewało różnic w markerach o niższej ekspresji. Tylko markery, które wykazały istotne różnice w przypadku leczenia THC, są przedstawione graficznie. Stwierdziliśmy, że w tkance limfatycznej okrężnicy THC zmniejszyło ekspresję DC CD86 w mLN i cLP, ale zwiększyło ekspresję CD80 w cLP DC. Ekspresja CD40 wzrosła w obecności THC w mLN DC, ale spadła z THC w cLP DC, a CD103 wzrosła w cLP DC. W makrofagach jelitowych ekspresja CD86 i Ly-6C została zmniejszona w makrofagach mLN i cLP, podczas gdy w cLP ekspresja CD80 i CD103 wzrosła w makrofagach, jak widać w DC ( Rysunek 7 B).
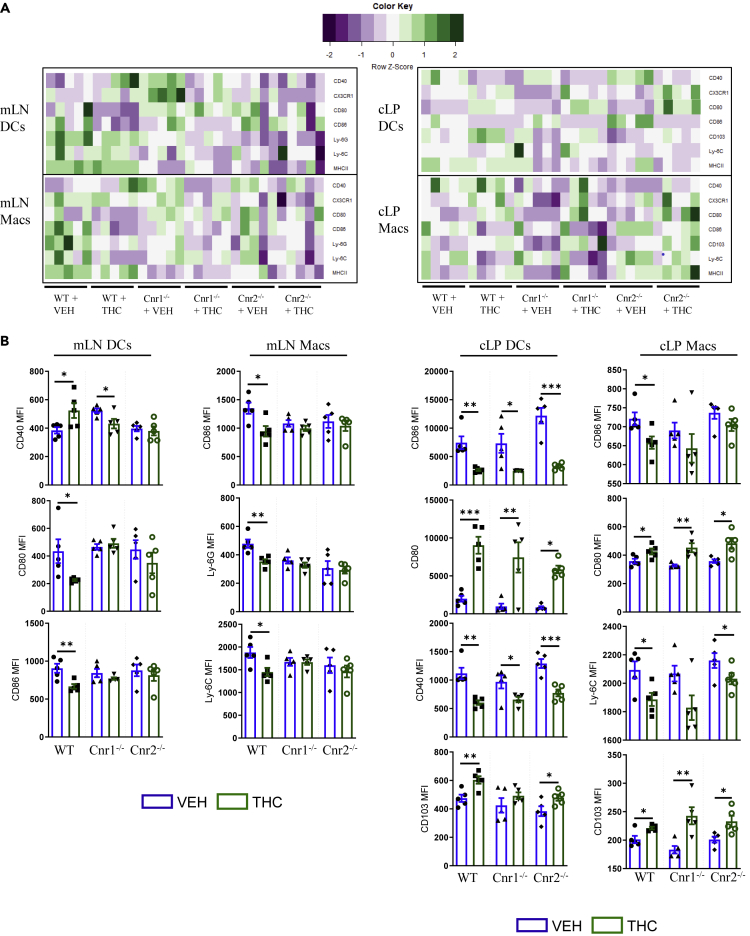
THC zmniejsza markery aktywacji APC poprzez receptory kannabinoidowe
Myszy WT, Cnr1 −/− i Cnr2 −/−, dopasowane pod względem wieku i płci, poddano eutanazji, a błonę właściwą jelita grubego (cLP), węzły chłonne krezkowe (mLNs), śledziony (spl), wątroby, płuca i mózg sprowadzono do zawiesiny pojedynczych komórek, a następnie poddano działaniu ex vivo VEH lub THC (10 μM) przez 18 godzin przed analizą markerów aktywacji komórek prezentujących antygen (APC) metodą cytometrii przepływowej.
(A) Mapa cieplna medianowych natężeń fluorescencji (w kolejności malejącej) CD40, CX3CR1, CD80, CD86, Ly-6G, Ly-6C i MHCII na makrofagach i komórkach dendrytycznych z tkanek pochodzących od myszy WT, Cnr1 −/− lub Cnr2 −/− leczonych VEH lub THC. Natężenie mapy cieplnej zostało znormalizowane dla każdego rzędu/markera zainteresowania.
(B) Kwantyfikacja wyników cytometrii przepływowej (n = 5).
Każdy symbol reprezentuje inną mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i są przedstawione jako średnia ± SEM. ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,005 metodą dwukierunkowej analizy wariancji z testem wielokrotnych porównań Tukeya.
W innych badanych tkankach zaobserwowano tendencję do zmniejszonej ekspresji wszystkich markerów u myszy Cnr2 −/− w porównaniu z myszami WT i Cnr1 −/− , co było najbardziej widoczne wśród komórek DC płuc ( rysunki S6 A–S6D). Co ciekawe, ani makrofagi, ani komórki DC w wątrobie nie wykazały zmiany markerów aktywacji z THC ( rysunek S6 A). Podobnie jak w jelitach, komórki DC płuc i śledziony wykazały spadek ekspresji CD86 pośredniczony przez CB2 w śledzionie ( rysunki S6 B i S6C). Ly-6C jest zmniejszone przez THC w komórkach DC płuc i mikrogleju, a CX3CR1 jest również zmniejszone w tych tkankach i w makrofagach śledziony ( rysunki S6 A–S6D). CD40 i Ly-6G są również zwiększone w mikrogleju z THC ( rysunek S6 D). Łącznie THC zmniejsza liczbę markerów aktywacji APC, zwłaszcza w barierach śluzówkowych jelit i płuc.
Aktywacja receptora kannabinoidowego 2 w komórkach mieloidalnych u myszy SCID pośredniczy w odporności jelita grubego na stan zapalny wywołany przez αCD40
W świetle danych dotyczących osłabiania zdolności stymulujących APC przez THC oraz faktu, że Tregs nie są mechanizmem, poprzez który THC zmniejsza produkcję IL-22 w okrężnicy, zainicjowaliśmy model αCD40 u myszy z niedoborem komórek T i B, aby zbadać, w jaki sposób modulacja komórek mieloidalnych przez THC wpływa na przebieg choroby. Podobnie jak u myszy WT BL/6, myszy SCID z niedoborem komórek T i B , którym podano αCD40, doświadczają ostrej utraty wagi, a leczenie VEH, THC lub THC plus antagonistami receptora CB AM251 (antagonista CB1) lub SR144528 (antagonista CB2) nie zmieniło wyniszczenia wywołanego przez αCD40 ( rycina S7 A). THC i THC + AM zmniejszyły stan zapalny okrężnicy, ale nie martwicze zapalenie wątroby ( ryciny 8 A–8C). Żadne leczenie nie zmniejszyło aktywności ALT w surowicy; jednak THC, THC + SR i THC + AM zmniejszyły aktywność AST w surowicy, co może sugerować zmniejszenie zaniku mięśni obserwowane w przypadku kannabinoidów ( rycina 8 D). Siedem dni po indukcji choroby, analizowano cLP DC i makrofagi, potwierdzając, że THC, THC + SR i THC + AM zwiększyły ekspresję CD103 DC, jak zaobserwowano u myszy WT ( ryciny 8 E i S7 B), ale na makrofagach tylko agonizm CB2 w grupie THC + AM zwiększył ekspresję CD103 przez makrofagi ( ryciny 8 E i S7 B). Komórki blaszki właściwej umieszczono na płytkach na noc i zebrano supernatanty, ujawniając, że leczenie THC i THC + AM zmniejszyło produkcję IL-23 i IL-1β, ale nie poziomy MCP-1, chociaż istniała tendencja do spadku ( rycina 8 F). U myszy naiwnych, którym podawano VEH lub THC przez 24 godziny, dane scRNA-seq wskazały, że podstawowa ekspresja Il1b i Ccl2 (MCP-1) nie uległa zmianie u myszy leczonych VEH i THC ( Rysunek S7 C). W tym modelu zapalenia okrężnicy wywołanego przez αCD40 z niedoborem komórek T, ILC3 są głównym źródłem okrężniczej IL-22, która nie uległa zmianie po jakimkolwiek leczeniu kannabinoidami ( Rysunek 8 G). Niemniej jednak, krążąca IL-22 została zmniejszona w grupach THC i THC + AM w porównaniu z grupami VEH i THC + SR w szczytowym momencie choroby i po zakończeniu badania ( Rysunek 8 H).
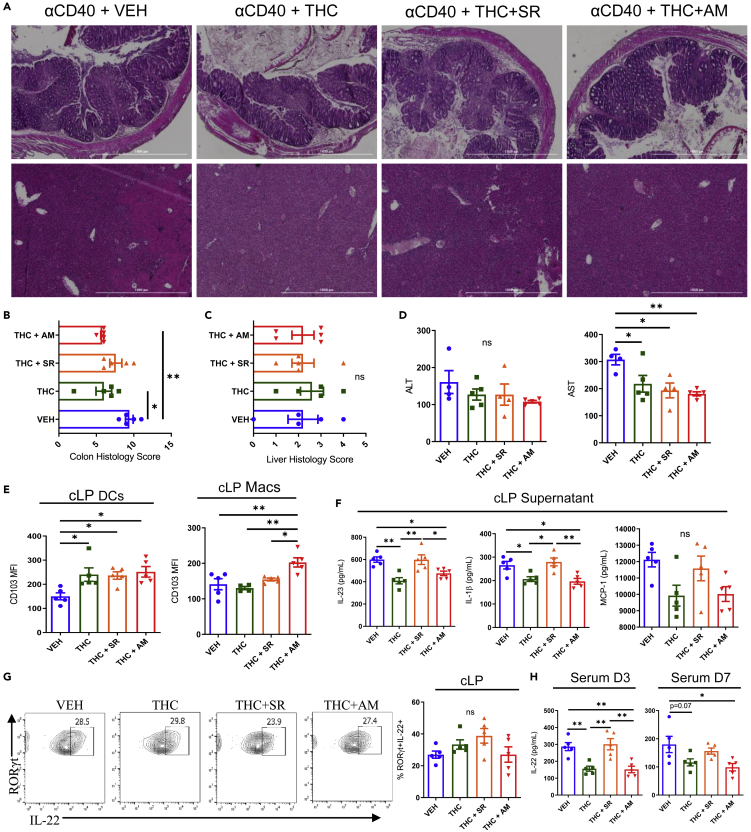
Aktywacja receptora kannabinoidowego 2 w komórkach mieloidalnych powoduje oporność jelita grubego na stan zapalny wywołany przez αCD40
Myszom SCID wstrzyknięto przeciwciała szczurze przeciwko myszom αCD40 (200 μg, klon FGK4.5 w PBS) i podawano im VEH, THC (10 mg/kg), THC+ SR144528 (THC+ SR, 10 mg/kg, oba) lub THC+ AM251 (THC + AM, 10 mg/kg, oba) codziennie przez 7 dni.
(A) Przykładowe obrazy H&E okrężnic (górny rząd, 4X) i wątroby (dolny rząd, 4X) u wskazanych myszy.
(B i C) (B) Ocena histologiczna obrazów H&E jelita grubego i (C) wątroby (n = 5).
(D) Aktywności ALT i AST mierzono w surowicy wskazanych myszy na zakończenie eksperymentu (n = 5).
(E) Mediana intensywności fluorescencji CD103 na komórkach DC i makrofagach z cLP myszy SCID , którym podano αCD40 i antagonistów VEH lub THC ± CB (n = 5).
(F) ELISA dla cytokin supernatantu cLP: IL-23, IL-1β i MCP-1 (n = 5).
(G) Wykresy konturowe cytometrii przepływowej komórek RORγt+ IL-22+ ILC3 w cLP wskazanych myszy po zakończeniu eksperymentu (n = 5).
(H) ELISA na obecność cytokiny IL-22 w surowicy w dniach 3 i 7 (n = 5).
Każdy symbol reprezentuje inną mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i są przedstawione jako średnia ± SEM. ∗p < 0,05, ∗∗p < 0,01 w dwukierunkowej analizie wariancji z testem wielokrotnych porównań Tukeya.
Rozwój zapalenia jelita grubego jest wieloczynnikowy i powstaje w wyniku braku równowagi między barierą gospodarza, komórkami odpornościowymi i komensalami ( Lynch i Pedersen, 2016 ). Najnowsze dowody sugerują, że IBD rośnie globalnie ( Coward i in., 2019 ; Kaplan i Ng, 2016 ), a ponieważ mało prawdopodobne jest, aby genetyka była winowajcą w skali czasowej, musimy wziąć pod uwagę interakcje między środowiskiem, komensalami i reakcją naszych komórek odpornościowych na nie, jako czynnik wywołujący chorobę. Modulacja interakcji jelitowych między dowolnymi aktorami może przechylić szalę w kierunku promocji lub ochrony przed IBD. Wiele badań wykazało, że kannabinoidy wykazują potencjał w przechylaniu tej skali w kierunku tolerancji w celu ochrony przed IBD ( Singh i in., 2012 ; Storr i in., 2009 ; Ke i in., 2016 ; Kimball i in., 2006 ). Jednak dane dotyczące działania przeciwbakteryjnego kannabinoidów u ludzi nie odzwierciedlają tego, co zaobserwowano w modelach mysich ( Ambrose i Simmons, 2019 ; Naftali i in., 2013 ; Lahat i in., 2012 ). W modelu AOM + DSS raka jelita grubego leczenie THC zapobiegło rozwojowi raka i zmniejszyło liczbę pronowotworowych komórek Th22 wytwarzających IL-22, które infiltrowały śródnabłonkowo. IL-22 zyskała uwagę ze względu na swoje właściwości rakotwórcze u pacjentów ludzkich i w modelach zwierzęcych raka jelita grubego, promując macierzystość raka poprzez aktywację STAT-3 ( Sun i in., 2015 ; Kryczek i in., 2014 ; Kirchberger i in., 2013 ). Nasze wyniki wykazały, że THC osłabiało nowotworową IL-22 w mikrośrodowisku kolonocytów działając poprzez CB2 na komórki hematopoetyczne; jednakże efekt ten nie był wewnętrzny dla Th22, ponieważ THC nie wpływał bezpośrednio na izolowaną generację komórek Th22 in vitro . Stymulacja zarówno Th22, jak i ILC3 do produkcji IL-22 jest regulowana przez prozapalne cytokiny, takie jak IL-1β, IL-6, MCP-1 i IL-23 z lokalnych komórek APC ( Parks i in., 2016 ; Manta i in., 2013 ; Dudakov i in., 2015 ). W związku z tym zastosowaliśmy model zapalenia jelita grubego anty-CD40 u myszy dzikiego typu oraz u myszy z niedoborem komórek T i B, aby zbadać, w jaki sposób THC może oddziaływać na adaptacyjny i wrodzony układ odpornościowy, redukując poziom IL-22 w modelu, w którym jest on patogenny dla proksymalnego odcinka jelita grubego, niezależny od mikrobioty i nie polega na substancjach chemicznych egzogennych w pośredniczeniu w postępie choroby ( Uhlig i in., 2006 ).
Wstrzyknięcie przeciwciał anty-CD40 powoduje ostrą wyniszczającą chorobę i zapalenie okrężnicy zależne od wydzielania cytokin zapalnych TNFα, IL-12 i IL-23 ( Eken i in., 2014 ; Longman i in., 2014 ; Uhlig i in., 2006 ). Odkryliśmy, że w modelu zapalenia okrężnicy anty-CD40 THC zmniejsza stan zapalny układowy i okrężnicy poprzez redukcję makroskopowej patologii jelitowej i krążących prozapalnych cytokin IFNγ, TNFα, IL-17A i IL-22. Stan zapalny układowy wywołany przez przeciwciała anty-CD40 został zmniejszony przez THC, co zmierzono na podstawie zmniejszenia splenomegalii i biomarkerów uszkodzenia wątroby AST i ALT. Leczenie THC u myszy anty-CD40 spowodowało wzrost Treg w cLP, co było pośredniczone przez THC działające bezpośrednio na DC w celu zwiększenia ekspresji CD103 i zmniejszenia ekspresji CD11b, przekształcając je w bardziej tolerogenne komórki ze zwiększoną ekspresją TGF-β1 potwierdzoną przez scRNA-seq. Działając poprzez CB2, wykazaliśmy, że THC moduluje fenotyp cLP DC w kierunku bardziej przeciwzapalnego stanu, charakteryzującego się zmniejszoną ekspresją CD80 i CD11b u myszy z zapaleniem jelita grubego anty-CD40 in vivo . Nasze dane scRNA-seq komórek mieloidalnych cLP i analiza cytometrii przepływowej ex vivo naiwnych DC pochodzących ze szpiku kostnego i DC pochodzących z cLP, mLN, śledziony i płuc, wszystkie wykazały zmniejszenie ekspresji CD86 z THC. Interesujące odkrycie z naszej analizy cytometrii przepływowej globalnych markerów aktywacji APC ujawniło, że podczas gdy THC polaryzowało DC w kierunku bardziej tolerogennego fenotypu ze zwiększoną ekspresją CD103 i zmniejszoną ekspresją CD86, THC działało również na DC mLN i mikroglej, zwiększając ekspresję CD40. Zwiększone sygnały CD40 na DC CD103+ w celu regulacji CCR7, powodując ich przesunięcie do mLN i apoptozę ( Barthels i in., 2017 ). Biorąc pod uwagę, że widzieliśmy, że THC zmniejsza ekspresję CCR7 w cLP i mLN, jest mało prawdopodobne, aby powodowało to apoptozę DC; jednak CD40 działa z wcześniej aktywowanymi antygenem komórkami T CD40L+ w celu współstymulacji w dodatniej pętli sprzężenia zwrotnego, gdy napotkają znane antygeny ( Cong i in., 2000 ). Zwiększenie ekspresji CD40 przez THC w mLN, ale zmniejszenie ekspresji innych markerów aktywacji sugeruje, że THC może pobudzać odporność jelitową na zidentyfikowane antygeny, ale zmniejszać jego skuteczność w stymulowaniu odpowiedzi immunologicznej na nowe antygeny w cLP, co może wyjaśniać, dlaczego kannabinoidy są nieskuteczne w usuwaniu modeli infekcji zapalenia okrężnicy, ale są skuteczne w modelach chemicznych ( Eisenstein i Meissler, 2015 ; Newton i in., 2009 ; Karmaus i in., 2011 ). Zwiększenie ekspresji CD40 przez THC może być podobnie mechanizmem kompensacyjnym wyjaśniającym zmniejszenie ekspresji CD80 i CD86.
Komórki DC w cLP mogą promować ekspansję komórek T regulatorowych. Kilka badań zbadało ten mechanizm, sugerując, że komórki DC, głównie CD103+ DC, regulują CCR7, aby migrować do mLN, gdzie wydzielają TGF-β1 i kwas retinowy, aby indukować komórki T regulatorowe ( Schulz i in., 2009 ). Nasze wyniki wykazały, że podanie THC myszom naiwnym i po zapaleniu okrężnicy wywołanym przeciwciałami anty-CD40 spowodowało zmniejszenie ekspresji CCR7 na komórkach DC zarówno w cLP, jak i mLN, co sugeruje, że THC zmniejsza migrację komórek DC między tkanką limfatyczną przewodu pokarmowego. Ponadto odkryliśmy, że komórki BMDC leczone THC i komórki z cLP myszy leczonych THC wykazywały zwiększone poziomy TGF-β1. Łącznie te dane wskazują, że w jelitach THC powoduje, że DC zamiast migrować do mLN i indukować Treg, pozostają w cLP, obniżają poziom markera aktywacji CD86, wydzielają TGF-β1 i zwiększają odsetek Treg oraz wpływają na inne lokalne typy komórek poprzez działanie przeciwzapalne TGF-β1. TGF-β1 jest krytyczny dla homeostazy jelitowej. Globalne myszy TGF-β1 −/− rozwijają spontaniczne zapalenie jelita grubego w wieku około 3–4 tygodni, a swoiste dla DC wyłączenie TGF-β1 powoduje spontaniczne zapalenie jelita grubego ( Ihara i in., 2017 ; Ramalingam i in., 2012 ). DC są ważnym źródłem i aktywatorem TGF-β1 w jelitach, niezbędnym do kontrolowania różnicowania Treg i Th17 ( Ramalingam i in., 2012 ) oraz homeostazy kolonocytów ( Ihara i in., 2016 ). Wpływ THC na DC wynika z obserwacji, że leczenie THC zmniejsza aktywność APC u myszy ( Karmaus i in., 2011 ) oraz in vitro w komórkach ludzkich ( Roth i in., 2015 ). Aby zbadać wkład komórkowy pośredniczony przez THC w redukcję nasilenia przeciwciał anty-CD40, najpierw zubożyliśmy Treg poprzez wstrzyknięcie przeciwciał anty-CD25, ujawniając, że redukcja stanu zapalnego układowego wywołanego przez przeciwciała anty-CD40 przez THC jest częściowo pośredniczona przez Treg, ponieważ u myszy pozbawionych Treg, którym podano THC, krążące IFNγ i TNFα wzrosły z powrotem do poziomów kontrolnych, a splenomegalia nie została zmniejszona. Badanie przeprowadzone na myszach Rag2 −/− wykazało, że adopcyjne przeniesienie Treg nie zmieniło stanu zapalnego układowego ( Bauché i in., 2018).); jednakże miarą, której użyli do wyciągnięcia tego wniosku, była masa ciała, która również nie uległa zmianie w naszym badaniu z THC, z Tregami lub bez nich, chociaż krążące biomarkery aktywności choroby i masa śledziony uległy zmianie. Niemniej jednak, zapalenie okrężnicy wywołane przez przeciwciała anty-CD40 u myszy WT nie jest łagodzone przez Treg indukowane przez THC, ponieważ wyczerpanie Tregów nie osłabiło redukcji wydzielania IL-17A lub ILC3 IL-22 przez THC. Dane dotyczące cytokin wykazały również, że THC nie zwiększało innych cytokin przeciwzapalnych podzbioru Th: IL-4, IL-10 lub IL-9, co sugeruje, że zbawienne właściwości THC tkwiły w jego modulacji APC.
Dane z myszy SCID z niedoborem komórek T i B , którym wstrzyknięto przeciwciało anty-CD40, potwierdziły to, co obserwowano u myszy WT — komórki APC z jelita grubego są stymulowane przez przeciwciało anty-CD40, aby wywołać prozapalne IL-23 i IL-1β, które są redukowane przez agonizm CB2, oraz MCP-1, który wykazywał nieistotny statystycznie trend spadkowy przy leczeniu THC i THC + AM251. Analiza scRNA-seq komórek mieloidalnych WT cLP wykazała podobny spadek Il1b i Ccl2 po leczeniu THC. Zmniejszenie stanu zapalnego jelita grubego u myszy SCID αCD40 nie było odzwierciedlone przez zmniejszenie stanu zapalnego układowego ocenianego na podstawie patologii wątroby i poziomów ALT, co potwierdza to, co zaobserwowaliśmy w badaniach zubożających Treg u myszy WT. THC i kombinacja THC i obu antagonistów zmniejszyły poziom krążącej AST, co sugeruje, że THC może zmniejszyć zanik mięśni w tym modelu, albo poprzez nadmiarowy efekt CB, albo poprzez GPR55. Głównym odkryciem z tego eksperymentu było to, że THC działało na oba wartownicze APC w okrężnicy, DC i makrofagi, zwiększając CD103 i zmniejszając mediatory prozapalne, które stymulują produkcję IL-22. Chociaż nasze dane cLP nie wykazują różnic w wydzielaniu ILC3, poziomy IL-22 w surowicy są znacząco zmniejszone zarówno w dniu 3, jak i 7 przy leczeniu THC i THC + AM, co potwierdza rolę CB2 w zmniejszaniu wydzielania IL-22.
W tym badaniu wykazujemy, że agonizm THC wobec CB2 jest mechanizmem, poprzez który jelitowe APC przechodzą fenotypową zmianę w kierunku bardziej przeciwzapalnego fenotypu charakteryzującego się zwiększoną ekspresją CD103 i zmniejszoną CD86. Te APC są mniej przygotowane do wydzielania prozapalnych mediatorów IL-23, IL-1β i MCP-1, zamiast tego zwiększając TGF-β1 w celu indukowania Treg w lokalnym mikrośrodowisku. Treg są ważne dla systemowego rozwiązania stanu zapalnego wywołanego przez αCD40, zdolnego do zmniejszenia stanu zapalnego wątroby i splenomegalii; jednak w jelicie grubym, to poprzez działanie THC na DC i makrofagi, THC może zmniejszyć patogenne poziomy IL-22, aby chronić gospodarza przed nadmiernym stanem zapalnym i rakiem. Tutaj analizujemy, w jaki sposób aktywacja CB2 może udoskonalić fenotyp komórek mieloidalnych, aby znieść stan zapalny i zapobiec zapaleniu okrężnicy i rozwojowi raka okrężnicy.
Ograniczenia badania
Wykorzystując model nowotworu de novo i modele zapalenia jelita grubego wywołanego chemicznie i przeciwciałami, badanie to opisuje mechanizmy, poprzez które THC, działając poprzez CB2 na komórki APC w jelitach, ogranicza wydzielanie prozapalnych cytokin, które w przeciwnym razie stymulowałyby onkogenne uwalnianie Th22 i ILC3 IL-22. Określenie, czy redukcja IL-22 u myszy leczonych THC jest jedynym czynnikiem zapobiegającym rozwojowi CRC, będzie pouczające i warte oceny w przyszłych pracach. Chociaż badanie to oceniało działanie zapobiegające nowotworom THC, badania oceniające wpływ THC na rozwinięte nowotwory byłyby klinicznie istotne.
źródło: https://pubmed.ncbi.nlm.nih.gov/32942172/
Pobierz PDF ![]()
{kind=link}
{kind=link}