
Rak jelita grubego jest poważnym problemem zdrowia publicznego. Niestety, obecnie nie istnieje żadna skuteczna opcja leczenia tego typu nowotworu złośliwego. Najbardziej obiecującym obecnie leczeniem raka jest immunoterapia, która jest również nazywana terapią biologiczną lub ukierunkowaną. Ten rodzaj terapii wzmacnia zdolność układu odpornościowego pacjenta do zwalczania nowotworu złośliwego. Jednak komórki nowotworowe mogą stać się oporne na immunoterapię i uniknąć nadzoru immunologicznego poprzez uzyskanie zmian genetycznych. Dlatego też konieczne są nowe strategie leczenia. W ostatniej dekadzie kilka raportów sugeruje skuteczność kannabinoidów i
ekstraktów
z Cannabis sativa w hamowaniu proliferacji nowotworów
in vitro i
in vivo , w tym nowotworów złośliwych jelit. Wykazano, że kannabinoidy modulują ścieżki zaangażowane w proliferację komórek, angiogenezę, zaprogramowaną śmierć komórek i przerzuty. Z tego powodu są proponowane jako terapia wspomagająca w przypadku wielu nowotworów złośliwych. O wiele mniej informacji istnieje na temat potencjału stosowania konopi w połączeniu z immunoterapią. W tym artykule analizujemy możliwość wykorzystania kannabinoidów w modulacji immunoterapii raka jelita grubego i omawiamy możliwe zalety i ograniczenia.
Obecnie rak jelita grubego (CRC) jest uważany za trzeci najbardziej śmiertelny i czwarty najczęściej wykrywany nowotwór na świecie ( 1 ). Pomimo obecności wysoce zaawansowanych technik przesiewowych, wskaźnik zapadalności stale rośnie na całym świecie ( 2 ). Szacuje się, że globalne obciążenie rakiem jelita grubego wzrośnie o 60% do ponad 2,2 miliona nowo zdiagnozowanych przypadków i 1,1 miliona zgonów do 2030 roku ( 3 ). Czynniki takie jak siedzący tryb życia, zwiększone spożycie alkoholu, tytoniu, czerwonego mięsa, predyspozycje genetyczne, przewlekłe procesy zapalne przewodu pokarmowego są czynnikami wyzwalającymi tego typu złośliwość ( 4 ). Wiadomo, że głównymi prekursorami CRC są polipy gruczolakowate. Szybkość transformacji tych polipów w raka wynosi ~0,25% rocznie. Gdy te zmiany mają wysoki stopień dysplazji i architekturę kosmków, ryzyko transformacji w nowotwór złośliwy wzrasta do 50% ( 5 ).
Zrozumienie patogenezy raka jelita grubego jest bardzo ważne dla wyboru właściwej terapii. Etiologia CRC jest złożona i obejmuje akumulację nabytych modyfikacji epigenetycznych i genetycznych, które przekształcają normalne komórki nabłonkowe w złośliwe. Klasyczny model progresji nowotworu nazywa się rozwojem sekwencji polipa-raka, która obejmuje trzy główne etapy. Pierwszym etapem jest tworzenie łagodnych nowotworów, takich jak gruczolaki i siedzące ząbkowane polipy. Drugi etap charakteryzuje się progresją łagodnych nowotworów w nowotwory bardziej zaawansowane histologicznie, a ostatni etap – ich transformacją w raka. Proces ten może trwać wiele lat bez wykazywania jakichkolwiek oznak i objawów. Kiedy CRC się rozwinie, może minąć jeszcze kilka lat, zanim zostanie zdiagnozowany. CRC jest spowodowany mutacjami w onkogenach, genach supresorowych nowotworu i genach zaangażowanych w mechanizmy naprawy DNA. Jedna z pierwszych mutacji występuje zazwyczaj w genie polipowatości gruczolakowatej jelita grubego (APC), który jest supresorem nowotworu, po czym następują mutacje w genach KRAS, TGF-β, BAX, BRAF i innych ( 6 ).
Większość przypadków CRC jest sporadyczna (70–80%), podczas gdy dziedziczne i rodzinne przypadki CRC stanowią odpowiednio około 5 i 25%. Sporadyczne nowotwory powstają z powodu mutacji punktowych, a molekularna patogeneza tych nowotworów jest bardzo niejednorodna. Dziedziczna grupa tego konkretnego nowotworu złośliwego jest spowodowana odziedziczonymi mutacjami i może być podzielona na dwie grupy: polipowatość i niepolipowatość. Typ polipowatości obejmuje głównie rodzinną polipowatość gruczolakowatą, która charakteryzuje się obecnością licznych, prawdopodobnie złośliwych polipów w jelicie grubym. Wariant niepolipowatości jest reprezentowany przez zespół Lyncha ( 7 ). Rodzinny CRC jest również spowodowany odziedziczonymi mutacjami i występuje rodzinnie bez obecności konkretnych dziedzicznych zespołów ( 8 ).
Ostatnio zaproponowano dwie molekularne klasyfikacje patologiczne oparte na szeroko zakrojonej analizie genomicznej i transkryptomowej CRC. Pierwsza z nich nazywa się The Cancer Genome Atlas (TCGA) i obejmuje trzy grupy: hipermutacje (13%), ultramutacje (3%) i niestabilność chromosomową (84%). Kategoria hipermutacji charakteryzuje się wysokim wskaźnikiem mutacji, wadliwą naprawą niedopasowań (dMMR) z dobrym rokowaniem, ale złym rokowaniem po nawrocie. Typ ultramutacji ma niezwykle wysoki wskaźnik mutacji z mutacją korekty epsilon polimerazy DNA i ogólnie dobrym rokowaniem. Większość CRC wyróżnia się niestabilnością chromosomową (CIN) z cechami niskiej częstości mutacji, ale wysoką częstością zmian liczby kopii somatycznych DNA. Druga klasyfikacja oparta na ekspresji genów nazywa się Consensus Molecular Subtypes (CMS) i obejmuje cztery grupy. CMS1 (14%) charakteryzuje się niestabilnością mikrosatelitarną (MSI), mutacją onkogenu BRAF i silną aktywacją immunologiczną. U pacjentów z tym podtypem zauważono słaby wskaźnik przeżycia po nawrocie. CMS2 (37%), zwany również kanonicznym, wykazuje wysoką niestabilność chromosomową i aktywację sygnalizacji WNT i MYC. CMS3 (13%), znany jako metaboliczny, ma liczne mutacje KRAS i deregulowane ścieżki metabolizmu. CMS4 (23%), zwany mezenchymalnym, jest opisany obecnością nacieku podścieliska, silnie ekspresjonowanymi genami mezenchymalnymi, aktywacją transformującego czynnika wzrostu beta, gorszym ogólnym i wolnym od nawrotów przeżyciem w porównaniu z pacjentami z innych grup ( 7 , 9 ). Klasyfikacje te dostarczyły informacji na temat właściwego doboru leczenia i rokowania pacjentów, co jest bardzo ważne dla trwających i przyszłych badań klinicznych.
Główne opcje terapeutyczne dostępne obecnie dla pacjentów z CRC to chirurgia, chemioterapia, immunoterapia, radioterapia. 5-letni wskaźnik przeżycia pacjentów z wczesnymi stadiami CRC wynosi prawie 90%. Ze względu na subtelne objawy, u ponad połowy pacjentów diagnozuje się chorobę, gdy rozwinęły się już zaawansowane nowotwory złośliwe. 5-letni wskaźnik przeżycia wynosi tylko 10% lub mniej, gdy pacjenci mają przerzuty ( 10 ).
Wśród nowych potencjalnych podejść terapeutycznych wykazano, że leczenie kannabinoidami i ekstraktami z Cannabis sativa jest skuteczne w hamowaniu wzrostu raka in vitro i in vivo ( 11 ). Roślina C. sativa zawiera fitokannabinoidy, terpenoidy, flawonoidy, kwasy tłuszczowe i inne cząsteczki. Kannabinoidy działają poprzez układ endokannabinoidowy, który składa się z receptorów, takich jak kannabinoid 1 (CB1), kannabinoid 2 (CB2), kanały potencjału receptora przejściowego podtypu waniloidowego 1 i 2 (TRPV1, TRPV2), receptory sprzężone z białkiem G 18, 55, 119 (GPR18, GPR55, GPR119), endokannabinoidy, takie jak 2-arachidonoiloglicerol i anandamid (2-AG, AEA) oraz enzymy odpowiedzialne za ich metabolizm. Głównymi enzymami biosyntetycznymi są NAPE-fosfolipaza D (NAPE-PLD) i lipaza diacyloglicerolowa (DAGL); głównymi enzymami degradacji są hydrolaza amidu kwasu tłuszczowego (FAAH) i lipaza monoacyloglicerolowa (MAGL). Główną funkcją układu endokannabinoidowego jest utrzymanie homeostazy ( 12 ). Receptor CB1 jest głównie ekspresowany w ośrodkowym układzie nerwowym, a receptor CB2, będący najbardziej rozpowszechnionym w układzie odpornościowym, występuje głównie w narządach obwodowych. Oba receptory są receptorami powierzchniowymi komórek sprzężonymi z białkiem G, które są sprzężone ze szlakami cyklazy adenylowej i kinazy białkowej cAMP A oraz szlakami MAPK i PI3K ( 13 ).
Znaczenie układu odpornościowego w raku jelita grubego
W przeszłości guzy były definiowane jako zbiór jednorodnych komórek nowotworowych. Agresywność nowotworu opisywano na podstawie jego cech kliniczno-patologicznych. Ostatnie postępy w immunologii i biologii molekularnej pozwoliły nam lepiej poznać podstawowe mechanizmy potencjału przerzutowego guzów. Wiele badań w tej dziedzinie poszerzyło wiedzę i podkreśliło znaczenie układu odpornościowego w regulacji wzrostu nowotworu. Głównymi graczami tego procesu są wrodzone komórki odpornościowe, takie jak neutrofile, makrofagi, komórki tuczne, eozynofile, komórki supresorowe pochodzące z mieloidu (MDSC) oraz adaptacyjne komórki odpornościowe, takie jak limfocyty T i B ( 14 , 15 ).
W ciągu ostatniej dekady wiedza na temat mikrośrodowiska guza (TME) stała się kluczem do zrozumienia złożonej, wieloetapowej tumorigenezy i opracowania nowych schematów leczenia i leków ( 16 ). Mikrośrodowisko raka obejmuje komórki rezydentne i nierezydentne, które są połączone ze sobą różnymi mediatorami, a każda z nich ma określoną funkcję. Komunikacja między tymi komórkami a komórkami nowotworowymi w ich otoczeniu zasadniczo reguluje los progresji guza. Komórki odpornościowe mogą hamować lub sprzyjać wzrostowi guza ( Tabela 1 ). Nowe badania przedkliniczne wykazały, że atypowe komórki nieprezentujące antygenu są najpierw atakowane przez wrodzony układ odpornościowy; następnie odpowiedź zapalna promuje tworzenie nowych naczyń krwionośnych i proliferację komórek nowotworowych. Niestety, guzy mogą włączać mechanizmy immunosupresyjne i unikać immunonadzoru gospodarza. Adaptacyjna odpowiedź immunologiczna wymaga identyfikacji antygenów nieswoistych poprzez komunikację między białkami a głównym kompleksem zgodności tkankowej komórek prezentujących antygen i receptorami komórek CD8+ i CD4+ T poprzez prezentację antygenu. Guzy mogą utracić swoją antygenowość z powodu nabytych wad w prezentacji antygenu lub mogą zostać zidentyfikowane jako własne ( 25 – 27 ).
Tabela 1.
Działanie pronowotworowe i przeciwnowotworowe komórek układu odpornościowego.
| Komórki odpornościowe | Rola w nowotworach (przeciwnowotworowa i pronowotworowa) | Odniesienia |
|---|---|---|
| Komórki dendrytyczne (DC) | Uwolnij cytotoksyczne cytokiny Prezentacja antygenu limfocytom T | ( 17 ) |
| Hamowanie funkcji komórek T poprzez ekspresję CTLA-4 Wspomaganie wzrostu i progresji guza | ||
| Limfocyty T (CD8+, CD4+) | Bezpośrednia liza komórek nowotworowych Uwalnianie cytotoksycznych cytokin | ( 18 ) |
| Uwalnianie cytokin sprzyjających powstawaniu raka | ||
| Komórki Treg | Hamuje przewlekły stan zapalny | ( 19 ) |
| Tłumienie odpowiedzi układu odpornościowego przeciwnowotworowego. Zwiększenie produkcji cytokin prozapalnych. | ||
| Makrofagi | Uwolnij cytotoksyczne cytokiny Prezentacja antygenu limfocytom T | ( 20 ) |
| Promowanie angiogenezy, proliferacji guza, chemotaksji, inwazyjności i przerzutów | ||
| Komórki supresorowe pochodzenia mieloidalnego (MDSC) | Ograniczony | ( 21 ) |
| Uwalniają immunosupresyjne mediatory molekularne. Tłumią funkcje komórek T. Rekrutują immunosupresyjne komórki odpornościowe. | ||
| Komórki NK | Uwalnia cytotoksyczne cytokiny Bezpośrednio zabija komórki rakowe | ( 22 , 23 ) |
| Granzym A ekspresowany na komórkach NK wspomaga rozwój raka poprzez nasilenie stanu zapalnego | ||
| Komórki tuczne | Hamuje wzrost komórek rakowych, zwiększa reakcję zapalną przeciwnowotworową | ( 24 ) |
| Wspomaganie wzrostu nowotworu poprzez stymulację neoangiogenezy, przebudowę tkanek i modulację odpowiedzi immunologicznej gospodarza |
Istnieją trzy fazy immunoedycji guza: eliminacja, równowaga i ucieczka ( Rysunek 1 ). W pierwszej fazie komórki układu odpornościowego eliminują komórki nowotworowe, które ekspresują białka powierzchniowe. W fazie równowagi niektóre komórki utrzymują się dzięki swojemu potencjałowi do kamuflowania cząsteczek powierzchniowych lub poprzez tłumienie makrofagów i komórek T poprzez ekspresję substancji takich jak PD-1/2 na komórkach prezentujących antygen. W ostatniej fazie niektóre komórki mogą uniknąć zabicia, co następnie prowadzi do ucieczki i proliferacji opornych klonów. Ponadto degradacja macierzy zewnątrzkomórkowej przez metaloproteinazy macierzy i nowe naczynia krwionośne utworzone w wyniku nieprawidłowej angiogenezy sprzyjają tworzeniu się przerzutów ( 15 ).
Rysunek 1.
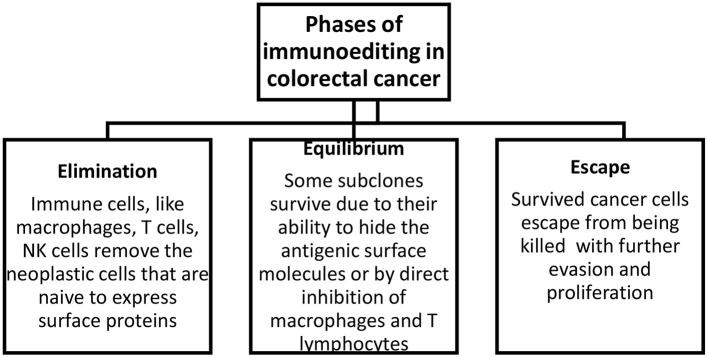
Fazy immunoedycji w raku jelita grubego. Eliminacja obejmuje usunięcie komórek nowotworowych, równowaga opisuje przeżycie frakcji przekształconych komórek, a ucieczka opisuje ucieczkę i proliferację tych komórek.
Jeśli chodzi o ekspresję receptorów kannabinoidowych w komórkach układu odpornościowego, wykazano, że receptory te są wyrażane zarówno w odporności adaptacyjnej, jak i wrodzonej. Na przykład receptory CB1, CB2 i GPR55 są wyrażane na komórkach NK, CB1, CB2 – na komórkach tucznych, limfocytach T – na komórkach B. Dlatego można postawić hipotezę, że fitokannabinoidy mogą wpływać na funkcjonowanie układu odpornościowego, regulować stan zapalny i posiadać działanie przeciwnowotworowe itp. ( 28 ).
Rola stanu zapalnego w karcynogenezie jelita grubego
Zapalenie odgrywa kluczową rolę w karcynogenezie jelita grubego i jest obecnie uważane za jedną z pojawiających się cech charakterystycznych raka ( 29 ). Lepsze zrozumienie CRC i zapalenia może doprowadzić do opracowania nowych biomarkerów nowotworowych i bardziej spersonalizowanych i skutecznych terapii. Wiadomo, że pacjenci cierpiący na przewlekłe schorzenia, takie jak choroba zapalna jelit, mają znacznie wyższe ryzyko rozwoju CRC ( 30 ). Zapalenie jest uważane za ważną siłę napędową nowotworów CRC związanych z zapaleniem jelita grubego, podczas gdy jego rola w przypadku nowotworów sporadycznych i dziedzicznych jest mniej jasna. Dowody wskazują, że niesteroidowe leki przeciwzapalne mogą zapobiegać rozwojowi CRC lub go opóźniać ( 31 ). Metaanaliza badań z randomizacją wykazała, że podczas obserwacji po 20 latach stosowania aspiryny przez 5 lat śmiertelność i częstość występowania CRC uległyby zmniejszeniu o 30–40% ( 32 ).
Na podstawie klasyfikacji CMS CRC, CMS1 i CMS4 są uważane za zapalne, przy czym pierwszy ma złe rokowanie po nawrocie, a drugi — najgorszy wskaźnik przeżycia. Ogólnie rzecz biorąc, stan zapalny odgrywa podwójną rolę w nowotworze. Celowanie w komórki złośliwe przez cytotoksyczne limfocyty T lub zmniejszanie niespecyficznego stanu zapalnego przez T-regs może prowadzić do odpowiedzi przeciwnowotworowej. Ten typ odpowiedzi nazywa się ochronną i jest związany z polaryzacją Th1 i niższym nawrotem CRC. Podtyp Th1 wytwarza IFN-γ i wzmacnia toksyczność komórkową, podczas gdy podtyp Th2 uwalnia IL-4 i wzmacnia humoralną odpowiedź komórek B. Najczęstszymi cytokinami prozapalnymi są TNF-α, IL-6, IL-12, IL-2, a najczęstszymi przeciwzapalnymi są IL-10, IL-4, IL-5, TGF-β i IFN-α ( Tabela 2 ). Komórki odporności wrodzonej i nabytej oraz inne komórki, takie jak fibroblasty, komórki mezenchymalne i perycyty, odgrywają ważną rolę w zapaleniu związanym z rakiem ( 57 ). Komunikacja między tymi komórkami odbywa się za pośrednictwem sieci cytokin produkowanych i wydzielanych przez komórki układu odpornościowego po stymulacji. Rola szlaku sygnałowego IL-10 pozostaje kontrowersyjna w przypadku CRC. Wyższy poziom IL-10 wiąże się z gorszym przeżyciem pacjentów, podczas gdy badania na zwierzętach pokazują, że pełni on rolę ochronną poprzez tłumienie stanu zapalnego ( 46 , 47 ). IL-6 jest aktywatorem szlaku sygnałowego STAT-3 i często występuje u pacjentów z CRC; i jest również powiązany z gorszym przeżyciem i zwiększonym ryzykiem nawrotu ( 37 , 57 , 58 ). Fibroblasty podścieliska, uzyskane z raka jelita grubego, wyprodukowały znaczne ilości IL-6. Ostatni z nich indukował angiogenezę guza poprzez zwiększenie produkcji VEGF ( 38 ). IL-6 ułatwia kolonizację przerzutową komórek raka jelita grubego. U myszy IL6-/- przerzuty komórek CT26 do wątroby zostały zmniejszone, a funkcja komórek CD8+ T uległa poprawie in vivo . Co więcej, myszy z niedoborem IL-6 skutecznie odpowiedziały na wstrzyknięcie anty-PD-L1 poprzez zahamowanie kolonizacji przerzutowej, podczas gdy u myszy IL6+/+ nie zaobserwowano tego efektu ( 39 ). IFN-γ jest wytwarzany przez komórki CD4+, CD8+ i NK i indukuje apoptozę komórek. Utrata jednej kopii tego interferonu u myszy Apcmin/+ wykazała znacznie szybszy postęp w kierunku gruczolakoraka jelita grubego. Komórki CRC mogą minimalizować przeciwnowotworowe efekty sygnalizacji interferonu przez łańcuch receptora interferonu typu I, co prowadzi do słabej odpowiedzi na inhibitory punktów kontrolnych anty-PD1 ( 44 ). Ekspresja TNF-α jest znacznie wyższa w raku jelita grubego niż w sąsiadującej prawidłowej tkance jelita grubego. Zwiększona ekspresja tej cytokiny silnie koreluje z bardziej zaawansowanymi nowotworami ( 59). Po stymulacji TNF-α zauważono wzrost onkogenu Metastasis-Associated in Colon Cancer 1 (MACC1) zarówno na poziomie mRNA, jak i białka. MACC1 indukuje proliferację, przeżycie i przerzuty komórek nowotworowych. Poziomy ekspresji tego onkogenu zostały zmniejszone przez obniżenie p65 NF-kB. Ponadto indukcję MACC1 utrudniało monoklonalne przeciwciało anty-TNF-α, adalimumab ( 34 ). TNF-α zwiększał poziomy cytokin prozapalnych, takich jak IL-6 i IL-8 in vitro na komórkach raka jelita grubego HT-29 ( 35 ). Inne badanie wykazało, że wpływ szczepionki peptydowej, AH1, na myszy z guzem jelita grubego CT26 spowodował skromne zahamowanie wzrostu guza, ale połączenie z F8-TNF drastycznie zwiększyło aktywność przeciwnowotworową. F8-TNF to białko fuzyjne przeciwciał, które dostarcza TNF do macierzy zewnątrzkomórkowej guza. Synergia między szczepionką peptydową a białkiem fuzyjnym TNF została wyjaśniona tym, że F8-TNF powoduje szybką martwicę krwotoczną guza i w rezultacie pozostawia niewielką ilość resztkowych komórek nowotworowych. Ponadto zauważono znaczący wzrost specyficznych dla AH1 komórek CD8+ T w guzach i drenujących węzłach chłonnych ( 60 ). IL-12 hamowała ruchliwość i inwazję ludzkich komórek raka jelita grubego (HRT18, HT29 i HT115), co sugeruje jej ważną rolę w przerzutach ( 41 ). IL-4 jest aktywnie uwalniana przez komórki macierzyste raka jelita grubego i nadaje guzom fenotyp odporny na śmierć. Neutralizacja IL-4 za pomocą przeciwciała znacząco uwrażliwia komórki nowotworowe na chemioterapię ( 49 ). Wczesna transgeneza IL-5 w mysim modelu CRC z zapaleniem jelita grubego zwiększyła ciężkość zapalenia jelita grubego, wywołała szybkość tworzenia polipów i w rezultacie większe obciążenie guzem ( 51 ). U pacjenta zgłoszono przypadek skrajnej eozynofilii spowodowanej przez rozsianego raka jelita grubego wytwarzającego IL-5 ( 61 ). TGF-β promuje przeżycie, inwazję i przerzuty komórek CRC ( 53 ). TGB-β w mikrośrodowisku guza zwiększa wykluczenie komórek T i hamuje wykopywanie fenotypu Th-1. Myszy z przerzutowym rakiem jelita grubego i zablokowaną ścieżką sygnałową TGF-β mają guzy wrażliwe na terapię anty-PD-1 anty-PD-L1. Natomiast myszy z odblokowaną sygnalizacją TGF-β wykazały ograniczoną odpowiedź na inhibitory punktów kontrolnych układu odpornościowego ( 54 ). Systemowe podanie IFN-α myszom z rakiem jelita grubego znacząco zahamowało wzrost guza i jego unaczynienie; indukowanej apoptozy komórek nowotworowych i komórek śródbłonka wątroby związanych z przerzutami ( 56 ).
Tabela 2.
Główne efekty cytokin pro- i przeciwzapalnych.
| Cytokiny | Miejsce produkcji | Ruchomości | Znaczenie dla karcynogenezy jelita grubego | Odniesienia |
|---|---|---|---|---|
| Cytokiny prozapalne | ||||
| TNF-alfa | Makrofagi, limfocyty T, komórki NK, komórki tuczne, eozynofile | Stymulacja stanów zapalnych, odporność na infekcje i nowotwory | TNF-α reguluje indukcję MACC1 poprzez podjednostkę NF-κB p65. Zwiększa poziom IL-6 i IL-8. | ( 33 ) ( 34 ) ( 35 ) |
| IL-6 | Limfocyty T, makrofagi | Stymulacja różnicowania komórkowego, stanu zapalnego i rozwoju limfocytów T efektorowych; indukuje syntezę białek ostrej fazy. | Powiązane z gorszym przeżyciem, zwiększone ryzyko nawrotu. Indukcja angiogenezy guza poprzez zwiększenie produkcji VEGF. IL-6 ułatwia przerzutową kolonizację komórek raka jelita grubego. | ( 36 ) ( 37 ) ( 38 ) ( 39 ) |
| IL-12 | Komórki dendrytyczne, makrofagi | Wspomaga rozwój odpowiedzi Th-1, zwiększa aktywność cytotoksyczną komórek NK i komórek T CD8+, wykazuje działanie antyangiogenne. | Hamowanie ruchliwości i inwazji komórek raka jelita grubego. | ( 40 ) ( 41 ) |
| IL-2 | Limfocyty T, komórki dendrytyczne | Przetwornik sygnału i aktywator transkrypcji (STAT5), wpływa na różnicowanie komórek pomocniczych T, aktywuje limfocyty cytotoksyczne | Ograniczone dane | ( 42 ) |
| IFN-γ | Komórki pomocnicze T (Th1), komórki NK | Reguluje równowagę Th1/Th2, wspomaga aktywację makrofagów, wzmacnia prezentację antygenu i migrację leukocytów, aktywuje STAT1. | Działanie przeciwnowotworowe poprzez spowolnienie postępu raka. | ( 43 ) ( 44 ) |
| Cytokiny przeciwzapalne | ||||
| IL-10 | Monocyty, limfocyty, komórki tuczne, makrofagi, komórki pomocnicze T (Th2), komórki regulatorowe T | Ograniczenie odpowiedzi immunologicznej gospodarza na patogeny, utrzymanie homeostazy tkankowej, zapobieganie rozwojowi chorób autoimmunologicznych; zmniejsza prezentację antygenu i fagocytozę, wzmacnia limfocyty T reg | Podwójna rola: tłumienie stanu zapalnego, co wiąże się z gorszym rokowaniem u pacjentów. | ( 45 ) ( 46 , 47 ) |
| IL-4 | Komórki tuczne, eozynofile, bazofile, limfocyty T | Reguluje równowagę Th1/Th2, indukuje alternatywną aktywację makrofagów i zmianę klasy immunoglobulin na IgE i IgG | Nadaje guzom fenotyp odporny na śmierć. Powoduje chemiooporność. | ( 48 ) ( 49 ) |
| IL-5 | Komórki pomocnicze T (Th2), komórki tuczne | Stymuluje proliferację komórek B i ich różnicowanie w komórki wydzielające Ig. | Pogorszyć przebieg choroby. | ( 50 ) ( 51 ) |
| TGF-β | Białe krwinki | Kontroluje proliferację komórek, różnicowanie i gojenie się ran; hamuje limfocyty B i aktywuje makrofagi; wspomaga różnicowanie limfocytów T. | Promuje przeżycie, inwazję i przerzuty komórek CRC. Zmniejsza odpowiedź na inhibitory punktów kontrolnych układu odpornościowego. | ( 52 ) ( 53 ) ( 54 ) |
| IFN-alfa | Komórki dendrytyczne plazmocytoidalne, makrofagi | Chemokineza i indukcja migracji komórek T, działanie przeciwwirusowe. | Wspomaga apoptozę komórek nowotworowych, hamuje angiogenezę i wzrost guza. | ( 55 ) ( 56 ) |
Aktualne metody leczenia raka jelita grubego
Optymalny sposób leczenia można wybrać, łącząc i analizując informacje na temat czynników związanych z nowotworem (lokalizacja nowotworu, obecność przerzutów, obecność biomarkerów itp.) oraz czynników związanych z pacjentem (rokowanie, choroby współistniejące itp.).
Pacjenci z rakiem jelita grubego z chorobą przerzutową otrzymują kombinację chemioterapii i immunoterapii. Chemioterapia pierwszego rzutu obejmuje fluoropirymidyny, takie jak kapecytabina i 5-fluorouracyl (5-FU) w monoterapii lub z leukoworyną (LV), oksaliplatyną (5-FU/LV/oksaliplatyna – FOLFOX), irynotekanem (5-FU/LV/irynotekan – FOLFIRI), kapecytabiną/LV/oksaliplatyną – CAPOX. Chemioterapia drugiego rzutu – FOLFOX lub CAPOX dla pacjentów opornych na irynotekan. Pacjentom opornym na kombinacje oksaliplatyny przepisuje się FOLFIRI lub irynotekan w monoterapii. Zwykle leczenie trwa do 6 miesięcy, ale czas trwania w znacznym stopniu zależy od indywidualnych przypadków ( 62 ).
Najczęstsze działania niepożądane chemioterapii w przypadku CRC to leukopenia, polineuropatia, biegunka, trombocytopenia, nadmierne wymioty, dysfunkcje wątrobowo-nerkowe i pogorszenie ogólnego stanu. Nasilenie działań niepożądanych jest zwykle większe u pacjentów w podeszłym wieku i u pacjentów z wcześniej istniejącymi chorobami współistniejącymi ( 63 ). Ze względu na obawy dotyczące toksyczności chemioterapia może nie być odpowiednia dla wielu pacjentów. Onkolodzy mogą nie zalecać tego typu leczenia ze względu na niektóre zaawansowane stadia chorób przewlekłych (niewydolność wątroby, nerek i serca) i słabą sprawność fizyczną ( 64 ).
Immunoterapia
Immunoterapia jest jedną z najbardziej obiecujących metod terapeutycznych dla pacjentów z rakiem jelita grubego ( 65 ). Terapia celowana zrewolucjonizowała leczenie raka. Immunoterapia jest rodzajem podejścia leczniczego, które pomaga układowi odpornościowemu w eliminacji guzów. Można ją podzielić na dwie główne grupy: aktywną (szczepionki) i pasywną (przeciwciała monoklonalne, adoptywna terapia komórkowa) ( Tabela 3 ). Ponadto niektóre terapie biologiczne mogą być szczególnie ukierunkowane na określone antygeny nowotworowe, podczas gdy inne działają niespecyficznie, wzmacniając naturalne odpowiedzi immunologiczne ( 75 ).
Tabela 3.
Zalety i wady środków immunoterapeutycznych.
| Immunoterapia | Zalety i wady | Status zatwierdzenia w CRC | Odniesienia |
|---|---|---|---|
| Szczepionki na cały guz | Składa się ze wszystkich znanych i nieznanych antygenów nowotworowych, łatwa produkcja | Niezatwierdzone | ( 66 ) |
| Niska immunogenność i skuteczność | |||
| Szczepionki peptydowe | Znana specyficzność antygenu związanego z guzem | Niezatwierdzone | ( 67 ) ( 66 ) |
| Niska skuteczność | |||
| Szczepionki wektorowe wirusowe | Specyficzny dla antygenu związanego z nowotworem, naturalnie immunogenny | Niezatwierdzone | ( 68 ) ( 66 ) |
| Indukcja burzy cytokinowej | |||
| Szczepionki z komórek dendrytycznych | Specyficzność antygenu związanego z nowotworem, generowanie własnej odpowiedzi immunologicznej | Niezatwierdzone | ( 69 ) |
| Przygotowanie jest kosztowne i czasochłonne | |||
| Adopcyjna terapia komórkowa | Wysoka specyficzność nowotworu, eliminacja konieczności wywoływania odpowiedzi immunologicznej | Niezatwierdzone | ( 70 ) ( 71 ) |
| Wysokie koszty, długi czas przygotowania, toksyczność zależna od celu | |||
| Immunoterapia oparta na przeciwciałach | Celowane szlaki immunosupresyjne, wzmocnienie odpowiedzi immunologicznej przeciwnowotworowej | Bewacyzumab Cetuksymab Panitumumab Ipilimumab Niwolumab Pembrolizumab | ( 72 ) ( 73 ) ( 74 ) |
| Toksyczność |
Istnieją pewne rodzaje szczepionek przeciwnowotworowych, które badano w leczeniu CRC, takie jak szczepionki na cały guz, peptydy, wektory wirusowe i komórki dendrytyczne (DC). Celem tych środków, podobnie jak każdej innej strategii immunizacji, jest wywołanie odpowiedzi immunologicznej przeciwnowotworowej, która wyeliminuje raka i zapewni organizmowi ciągły nadzór w celu ochrony przed jego nawrotem.
Szczepionki na cały guz
Niektóre zalety pracy ze szczepionkami na cały guz to: są łatwe w produkcji i składają się ze wszystkich znanych i nieznanych antygenów nowotworowych. Natomiast najistotniejszą wadą tych szczepionek jest bardzo niska immunogenność, która może być skierowana do normalnych komórek, a w rezultacie niska skuteczność. Podjęto kilka podejść w celu zwiększenia immunogenności szczepionek na cały guz. Przeprowadzono badanie z zakażeniem wirusem choroby Newcastle, które wykazało 98% 2-letni wskaźnik przeżycia u pacjentów z wyciętym rakiem jelita grubego w porównaniu z 67% u pacjentów, którzy otrzymali szczepionkę na cały guz w połączeniu ze szczepionką Bacillus Calmette-Guérin (BCG). Wyniki sugerują, że immunogenność tych związków została poprawiona ( 27 ).
Szczepionki peptydowe
Szczepionki peptydowe są bardziej specyficzne dla antygenu związanego z guzem, ale ich skuteczność jest nadal znacznie niska ze względu na niewielką ilość odpowiedzi komórek T. Badanie fazy I/II przeprowadzone u pacjentów z rakiem jelita grubego wykazało, że połączenie szczepionki p53 z interferonem alfa zwiększyło ilość interferonu gamma ( 76 ). Następny typ szczepionki to szczepionki wektorowe wirusowe. Są specyficzne dla antygenu związanego z guzem i naturalnie immunogenne. Wadą ich stosowania jest ich zdolność do wywoływania burzy cytokinowej. Najczęściej stosowanymi wirusami w raku jelita grubego są adenowirusy, pokswirusy i alfawirusy. Większość z tych szczepionek jest ukierunkowana na antygen karcinoembrionalny (CEA), białko wyrażane przez raki jelita grubego. Dane przedkliniczne pokazują, że rekombinowany wirus ospy wyrażający CEA (rV-CEA) może wzmacniać adaptacyjne i wrodzone odpowiedzi immunologiczne u myszy. Ponadto hamował proliferację gruczolakoraka jelita grubego w modelach zwierzęcych. Jednakże badania kliniczne przeprowadzone na pacjentach w zaawansowanym stadium raka jelita grubego wykazały brak odpowiedzi ( 77 ). Szczepionki z komórek dendrytycznych charakteryzują się specyficznością antygenu związanego z guzem i generowaniem własnej odpowiedzi immunologicznej organizmu. Negatywnymi aspektami są wysokie koszty i bardzo czasochłonny proces przygotowania. Po całkowitym wycięciu przerzutów CRC do wątroby, badanie kliniczne szczepionki fazy II wykazało mniej i opóźnione nawroty w grupie szczepionej w porównaniu z grupą obserwacyjną ( 78 ). Wyniki szczepionek DC są bardzo zachęcające, a wkrótce ich skuteczność może zostać znacznie poprawiona.
Terapia Adopcyjnego Transferu Komórek
Adopcyjna terapia transferu komórek to kolejny rodzaj immunoterapii. Głównymi zaletami tego typu leczenia są eliminacja konieczności wytworzenia odpowiedzi immunologicznej i wysoka swoistość guza. Z kolei pewnymi wadami są wysokie koszty, długi czas przygotowania i toksyczność zależna od celu. W tej terapii autologiczne komórki T są pobierane z guza, węzłów chłonnych lub krwi obwodowej pacjenta i modyfikowane ex vivo poprzez ich ekspansję i dodanie niektórych cząsteczek współstymulujących i cytokin. Następnie przeprowadza się bierny transfer tych komórek T do gospodarza w celu bezpośredniego zniszczenia guza. Najnowszym odkryciem tego typu biernej immunoterapii jest opracowanie zmodyfikowanych komórek T, które wyrażają chimeryczne receptory antygenowe specyficznie dla antygenu karcinoembrionalnego ( 79 ). Badanie fazy I przeprowadzone u pacjentów z rakiem jelita grubego opornym na standardowy schemat protokołu leczenia przy użyciu autologicznych komórek T zmodyfikowanych w celu wyrażenia receptora komórek T CEA myszy wykazało znaczący spadek stężenia CEA w surowicy u wszystkich trzech pacjentów; u jednego z nich wykazano odpowiedź kliniczną w postaci regresji przerzutów w wątrobie i płucach. Jednocześnie u tych pacjentów wystąpiło przejściowe zapalenie jelit ( 80 ). Niedawno opublikowane wyniki badania przypadku u pacjentów z zaawansowanym rakiem jelita grubego wykazały znaczącą odpowiedź kliniczną na połączenie kapecytabiny i adopcyjnego transferu komórek (komórek αβT i komórek NK) przepisanych po laparoskopowej resekcji raka jelita grubego i niektórych przerzutów do wątroby. Dwa tygodnie po laparoskopii zaobserwowano drastyczny wzrost poziomu CEA. Adopcyjny transfer komórek pozwolił na obniżenie poziomu CEA w surowicy, ostatecznie doprowadzając go do normy. Zaobserwowano zauważalną redukcję wielkości nieoperacyjnych przerzutów do wątroby. Podczas badania kontrolnego po 19 miesiącach nie odnotowano progresji ani nawrotu, a poziomy CEA utrzymywały się w granicach normy ( 81 ).
Immunoterapia oparta na przeciwciałach
Wysoce specyficzne przeciwciała monoklonalne są bardzo skuteczne w leczeniu raka od dziesięcioleci. Białka przeciwko receptorowi naskórkowego czynnika wzrostu (EGFR) i czynnikowi wzrostu śródbłonka naczyniowego (VEGF) w połączeniu z chemioterapią wykazały lepsze wyniki leczenia złośliwego CRC (mCRC). Środki anty-EGFR, takie jak Cetuximab lub Panitumumab, w monoterapii lub w połączeniu z lekami cytotoksycznymi są przepisywane tylko wtedy, gdy nie ma mutacji KRAS ( 62 ). Bevacizumab, humanizowane przeciwciało monoklonalne przeciwko VEGF, hamuje wzrost guza i angiogenezę, a także moduluje układ odpornościowy gospodarza poprzez zwiększenie populacji komórek B i T ( 82 ).
Dramatyczną skuteczność immunoterapii opartej na przeciwciałach udowodniono przy użyciu innego typu przeciwciał monoklonalnych (mAbs) znanych jako inhibitory punktów kontrolnych (ICI). Wykazano, że obecnie stosowane ICI zapewniają znaczące odpowiedzi kliniczne u pacjentów z mCRC, szczególnie z typem niedoboru naprawy niedopasowania/wysokiej niestabilności mikrosatelitarnej (dMMR-MSI-H). Celują one w hamujące receptory immunologiczne: programowaną śmierć komórki 1 (PD-1) i jej ligand PD-L1, cytotoksyczny antygen związany z limfocytami T 4 (CTLA-4). Ten ostatni jest wyrażany w naiwnych limfocytach T, efektorowych limfocytach T i regulatorowych limfocytach T (T-regs). Stymuluje on dezaktywację T-regs poprzez wiązanie się z komórkami prezentującymi antygen. Receptor PD-1 jest obecny w limfocytach CD4, CD8, komórkach NK, MDSC, T-regs i limfocytach B. Razem ze swoim ligandem, ten receptor powoduje wyczerpanie komórek T poprzez minimalizowanie naciekających guz limfocytów i proliferacji komórek T. W konsekwencji, guzy nabywają immunooporności. Środki anty-CTLA-4 i anty-PD-1 aktywują komórki T i powodują silniejszą odpowiedź przeciwnowotworową ( 83 ). Guzy CRC dMMR-MSI-H mają 20 razy większy ładunek mutacji niż guzy z niestabilnością mikrosatelitarną o niskiej sprawności naprawy niezgodności (pMMR-MSI-L). Ponadto są bardziej naciekane przez TIL, makrofagi i mają podwyższone poziomy cytokin stymulujących układ odpornościowy w porównaniu z pMMR-MSI-L. Ten ostatni ma mniej skuteczną odpowiedź na ICI i gorsze rokowanie ( 84 ).
W sierpniu 2020 r. istniały trzy zatwierdzone przez FDA ICI, które były stosowane u pacjentów z dMMR-MSI-H mCRC. Pierwszym z nich był Nivolumab, lek przeciwko PD-1 zatwierdzony przez FDA w lipcu 2017 r. po pomyślnych wynikach badania fazy II CheckMate 142 dotyczącego leczenia drugiej linii pacjentów z dMMR-MSI-H mCRC, u których wystąpił postęp choroby podczas leczenia oksaliplatyną, fluoropirymidyną i irynotekanem. W badaniu tym zgłoszono, że po 12 miesiącach obserwacji obiektywny wskaźnik odpowiedzi wystąpił u 31% pacjentów i 69% osób z grupy kontrolnej. 12-miesięczne przeżycie bez progresji (PFS) wyniosło 50%, a całkowite przeżycie (OS) 73%. Najczęstszymi działaniami niepożądanymi związanymi z terapią były świąd, wysypka, biegunka i zmęczenie. Mutacje BRAF, KRAS i ekspresja PD-L1 nie miały wpływu na odpowiedź na przepisaną terapię celowaną ( 85 ).
Drugim ICI zatwierdzonym przez FDA w maju 2017 r. był Pembrolizumab, substancja przeciwko PD-1, której skuteczność została udowodniona w pierwszej fazie badania Keynote 016. Początkowo wykazano, że pacjenci z mCRC dMMR-MSI-H doświadczyli 40% wskaźnika odpowiedzi (RR), podczas gdy pacjenci z pMMR-MSI-L – 0% RR. Później udokumentowano, że 2-letni PFS wynosił 53% w pierwszej grupie. Ciężkie działania niepożądane wystąpiły tylko u 14% pacjentów, w tym trombocytopenia, leukopenia i zapalenie trzustki. Ta lecznicza opcja monoterapii została przepisana pacjentom z dMMR-MSI-H mCRC, których stan pogorszył się w trakcie lub po terapii oksaliplatyną, fluoropirymidyną i irynotekanem.
Następnym podejściem immunoterapeutycznym zatwierdzonym przez FDA w przypadku opornego CRC, który rozwinął się podczas terapii oksaliplatyną, fluoropirymidyną i irynotekanem, było połączenie niwolumabu z ipilimumabem (środek anty-CTLA-4). Zatwierdzenie zostało przyznane w lipcu 2018 r., po raporcie wyników badania fazy II CheckMate 142. Podczas obserwacji po 13,4 miesiącach obiektywny wskaźnik odpowiedzi wyniósł 54,7%, przy częściowej odpowiedzi −51,3%, całkowitej odpowiedzi −3,4%, a wskaźnik kontroli choroby przez 3 miesiące lub dłużej −80%. PFS po 12 miesiącach wyniósł 71%, OS −85%. Trzynaście procent pacjentów było zmuszonych przerwać leczenie z powodu działań niepożądanych związanych z lekiem. Ta kombinacja wykazała lepszą skuteczność niż monoterapia anty-PD-1. Jednakże działania niepożądane stopnia 3–4 były bardziej widoczne w przypadku terapii skojarzonej w porównaniu z leczeniem jednym lekiem i wynosiły odpowiednio 32–20% ( 86 ).
W przypadku pacjentów z pMMR-MSI-L należy przeprowadzić więcej badań nad schematami immunoterapeutycznymi. Istnieje potrzeba znalezienia leków, które będą ukierunkowane na odpowiedź immunologiczną, a także będą promować naciek komórek T. Ze względu na niskie obciążenie mutacjami i neoantygenami trudno jest osiągnąć te cele. Obecne schematy obejmują radioterapię, chemioterapię i substancje antyangiogenne w celu zwiększenia aktywacji immunologicznej, zabijania komórek nowotworowych i podwyższenia antygenów nowotworowych. Później leczenie można łączyć z ICI i innymi lekami biologicznymi. Obecnie trwają pewne badania kliniczne, które oceniają skutki chemioterapii z zastosowaniem terapii anty-PD-1, anty-PD-L1 i zewnętrznej radioterapii wiązką lub ablacji częstotliwości radiowej ( 87 ). Trwające badanie NCT01633970 fazy Ib oceniało skuteczność atezolizumabu (anty-PD-L1) i bevacizumabu plus FOLFOX; wykazało ono OS na poziomie 7% i stabilizację choroby u 64% pacjentów. Innym dobrze przebadanym podejściem jest połączenie inhibitorów kinazy białkowej aktywowanej mitogenem (MEK), takich jak Cobimetinib i Atezolizumab. Inhibitory MEK mogą dodatkowo uwrażliwić MSS mCRC na terapię celowaną. Badanie kliniczne fazy Ib ( NCT01988896 ) oceniło to połączenie u pacjentów z opornym na leczenie mutantem KRAS CRC i pMMR-MSI-L CRC i wykazało RR wynoszący 17%, gdzie pięciu pacjentów z 23 miało stabilną chorobę, a u czterech pacjentów rozwinęła się PR. Nie odnotowano żadnych zaawansowanych działań niepożądanych związanych z terapią. Później włączono 84 pacjentów, a wyniki zaktualizowano. RR wyniosło 8%, wskaźnik kontroli choroby -−31%. 6-miesięczne PFS i 12-miesięczne OS wyniosły odpowiednio 27 i 51%. To podejście jest bardzo obiecujące, ponieważ pokazuje, że inhibitory MEK mogą zwiększyć odpowiedź na immunoterapię u pacjentów z MSS mCRC. Podczas trwającego badania NCT03406871 , w którym połączono niwolumab i regofarenib (inhibitor wielokinazowy), przedstawiono obiecujące wyniki ; u 18 z 19 pacjentów wystąpiła obiektywna odpowiedź guza (siedem z nich to MSS CRC, 11 — rak żołądka MSS i 1 — MSI-H CRC). Nadal konieczne są bardziej spersonalizowane podejścia do leczenia pMMR-MSI-L ( 84 ).
Znaczenie ECS dla CRC
ECS aktywnie reguluje homeostazę jelit. Wszystkie składniki ECS są silnie wyrażone w tkance jelitowej, co oznacza, że ten system bezpośrednio wpływa na prawidłowe funkcjonowanie układu żołądkowo-jelitowego. Receptory CB1 i CB2 są wyrażone w zdrowym nabłonku okrężnicy, podśluzówkowym splocie mięśniowo-jelitowym i mięśniach gładkich, komórkach plazmatycznych w blaszce właściwej; receptor CB2 jest również obecny na makrofagach jelitowych ( 88 , 89 ). Receptor TRPV1 jest wyrażany na włóknach nerwowych okrężnicy ( 90 ). Receptor GPR55 jest obecny w błonie śluzowej i warstwie mięśniowej okrężnicy ( 91 ). Endokannabinoidy, 2-AG i AEA są również obecne w zdrowej tkance okrężnicy ( 92 ). Główne enzymy degradujące endokannabinoidy, enzymy FAAH i MAGL są rozmieszczone na gruczołach nabłonka okrężnicy, blaszce właściwej i splocie mięśniowo-jelitowym. Enzymy biosyntezy NAPE-PLD i DAGL występują na mięśniach gładkich jelita grubego, blaszce właściwej i gruczołach nabłonkowych; DAGL występuje również na splocie mięśniówkowym jelita ( 89 ).
Aby zrozumieć rolę ECS w jelitach, ważne jest rozróżnienie skutków zwiększonego i zmniejszonego tonu kannabinoidowego w układzie żołądkowo-jelitowym. Ogólnie rzecz biorąc, antagoniści receptora CB1 zmniejszają ton kannabinoidowy w jelitach i prowadzą do wymiotów, biegunki, zwiększonego opróżniania żołądka i pasażu żołądkowo-jelitowego. Natomiast agoniści receptora CB1 i CB2, a także inhibitory MAGL i blokery FAAH prowadzą do zwiększenia tonu kannabinoidowego jelit poprzez zmniejszenie wymiotów, wydzielania kwasu żołądkowego i opróżniania żołądka, a także zmniejszenie nadruchliwości, biegunki i bólu trzewnego ( 93 ). Wyciszenie receptora CB1 przez selektywnego antagonistę receptora CB1 AM251 u myszy ApcMin/+ doprowadziło do zwiększenia liczby polipów jelitowych, podczas gdy aktywacja receptora CB1 spowodowała śmierć komórek nowotworowych. Natomiast wyciszenie receptora CB2 nie wykazało żadnego wpływu na wzrost polipów ( 94 ).
Składniki ECS są znacząco rozregulowane w CRC. Endokannabinoidy (2-AG i AEA) były 3-krotnie wyższe w gruczolakach i 2-krotnie wyższe w CRC w porównaniu do prawidłowej błony śluzowej jelita grubego ( 92 ). Ekspresja receptora CB1 jest zmniejszona w CRC ( 95 ). Ekspresja receptora CB2 jest zwiększona w CRC i jest uważana za zły czynnik prognostyczny w tym typie raka ( 96 ). Poziomy FAAH i MAGL były również zwiększone u pacjentów z CRC ( 97 ). ECS jest bardzo ważnym czynnikiem patogenezy CRC, co sugeruje potencjalny wpływ kannabinoidów na tę chorobę.
Roślina lecznicza , która ostatnio zyskała wiele uwagi w dziedzinie leczenia raka, to Cannabis sativa . Wiele eksperymentów in vitro i in vivo wykazało, że kannabinoidy i ekstrakty z konopi hamują proliferację, stymulują apoptozę i autofagię, tłumią angiogenezę i przerzuty ( 98–100 ). Głównymi aktywnymi kannabinoidami odpowiedzialnymi za te efekty są kannabigerol (CBG), kannabidiol (CBD) i tetrahydrokannabinol (THC). Wykazano, że CBG aktywował apoptozę, stymulował produkcję ROS, zwiększał mRNA CHOP i hamował wzrost komórek w komórkach CRC (Caco-2, HCT-116) ( 101 ). Stwierdzono, że hamujący wpływ CBG na żywotność komórek raka jelita grubego zależał od czasu. W wyciszonych komórkach TRPM8 hamujący wpływ CBG na wzrost komórek był wyraźnie tłumiony w porównaniu z komórkami niewyciszonymi. Indukcję apoptozy wykazano poprzez wzrost aktywności kaspaz 3 i 7, obecność fragmentów DNA, wzrost ekspresji CHOP. W tym samym artykule wykazano, że CBG (3 lub 10 mg/kg) hamowało wzrost guzów ksenoprzeszczepowych (HCT-116) w modelu myszy o 45,3% i chemicznie indukowało karcynogenezę jelita grubego w modelach za pomocą azoksymetanu (AOM), w którym CBG w stężeniu 5 mg/kg całkowicie hamowało powstawanie nieprawidłowych ognisk krypt (ACF), zmniejszało liczbę guzów o połowę i nie wpływało na powstawanie polipów ( 101 ).
Wykazano również, że CBD ma działanie antyproliferacyjne w modelach raka jelita grubego. W niektórych badaniach in vitro CBD chroniło DNA przed stresem oksydacyjnym, podnosiło poziom endokannabinoidów i hamowało proliferację komórek raka jelita grubego za pośrednictwem receptorów CB1, TRPV1, PPAR-γ ( 102 ). Selektywni antagoniści rimonabant i AM251 (antagonista CB1R), kapsazepina (antagonista TRPV1R), GW 9662 (antagonista receptora PPAR-γ) hamowali działanie antyproliferacyjne CBD. Chemoprewencja CBD została potwierdzona przy użyciu modeli in vivo raka jelita grubego wywołanego AOM. CBD (1 mg/kg) zmniejszyło ACF o 67%, liczbę guzów o 66% i polipów o 57%. Gdy stężenie wzrosło do 5 mg/kg, zapobiegało jedynie tworzeniu się polipów. Efekt ten był spowodowany aktywacją kaspazy-3 i zmniejszeniem fosforylowanej formy białka Akt ( 102 ). W innym badaniu wykazano proapoptotyczny efekt CBD w komórkach CRC (HCT-116, DLD-1) i zasugerowano, że jest on wynikiem aktywacji Noxa, zwiększenia produkcji ROS i indukcji stresu siateczki śródplazmatycznej. Gdy poziomy Noxa zostały stłumione przez siRNA, ekspresja markerów apoptozy uległa znacznemu zmniejszeniu. Podobnie, po zablokowaniu produkcji ROS, poziom Noxa został zmniejszony. CBD indukował apoptozę w sposób zależny od Noxa-ROS ( 103 ). Ponadto, podczas stosowania raka jelita grubego indukowanego linią komórkową CT26 u myszy, CBD w stężeniach 1 i 5 mg/kg wykazywało działanie przeciwangiogenne i przeciwprzerzutowe poprzez hamowanie VEGF, przy czym ta druga dawka była skuteczniejsza. U zwierząt otrzymujących CBD zaobserwowano istotny wzrost aktywności enzymów antyoksydacyjnych, w tym SOD, GPX, GR, TAC i spadek MDA ( 104 ).
Badano również wpływ pełnych ekstraktów botanicznych, takich jak botaniczna substancja lecznicza o wysokiej zawartości CBD (BDS), na raka jelita grubego. Takie ekstrakty są zazwyczaj przygotowywane z kwiatów konopi, które są bogate w CBD, lub izolat CBD jest dodawany (wzbogacany) do określonego stężenia. Postawiono hipotezę, że inne składniki ekstraktów z roślin konopi mogą działać synergicznie z CBD i mogą być przydatne z terapeutycznego punktu widzenia. Wykazano, że CBD BDS ma znaczące właściwości antyproliferacyjne na komórki rakowe (HCT-116, DLD-1), podczas gdy zdrowe komórki nabłonka jelita grubego nie zostały dotknięte. Nie odnotowano różnicy w sile i skuteczności między CBD BDS a CBD, gdy stosowano te same dawki (0,3–5 μM). Efekty CBD BDS były neutralizowane przez selektywnych antagonistów receptorów CB1 i CB2. CBD BDS miało wyraźniejsze powinowactwo do receptorów CB1 i CB2 niż czysty CBD. Badania in vivo wykazały, że zastosowanie chemicznie indukowanej karcynogenezy przez AOM, ekstraktu z C. sativa o wysokiej zawartości kannabidiolu, zahamowało ACF o 86%, polipy o 79% i tworzenie się guzów o 40%. W modelach ksenoprzeszczepu CBD BDS znacząco zmniejszyło objętość guza, ale nie zaobserwowano różnicy w rozwoju guzów po 1 tygodniu leczenia ( 105 ).
Wykazano, że THC indukuje apoptozę w komórkach raka jelita grubego poprzez aktywację receptorów CB1 i hamowanie PI3K-AKT, kaskady RAS-MAPK i aktywacji BAD. Komórki raka jelita grubego (SW480, HCT-15, HT29, Caco-2, HCT-116 i SW620) wystawione na działanie THC (2,5–12,5 μM) skutkowały dawkozależnym zmniejszeniem przeżywalności komórek. Natomiast mniejsze stężenia od 100 nM do 1 μM nie miały zauważalnego wpływu na proliferację i przeżywalność komórek raka jelita grubego. THC zwiększało poziomy kaspazy-3 i PARP (substratu kaspazy-3). THC powodowało defosforylację i aktywację BAD ( 106 ). Potencjał przeciwnowotworowy kannabinoidów w CRC podsumowano w Tabeli 4 .
Tabela 4.
Potencjał przeciwnowotworowy kanabinoidów w raku jelita grubego.
| Fitokannabinoidy | Typ modelu | Działanie przeciwnowotworowe/mechanizm działania | Odniesienia |
|---|---|---|---|
| CBG | in vitro (Caco-2, HCT116) in vivo (samice myszy grasicy, ksenograft-HCT116; model raka jelita grubego wywołanego azoksymetanem) | Proapoptotyczne, antyproliferacyjne; pobudza produkcję ROS, zwiększa mRNA CHOP, zwiększa poziom aktywności kaspazy 3, 7; w guzach ksenoprzeszczepionych – zmniejsza wzrost guza, w guzach AOM – całkowicie hamuje ACF, zmniejsza liczbę guzów | ( 101 ) |
| CBD | (Caco-2, HCT116) in vivo (samce myszy ICR, CRC wywołane AOM) | Antyproliferacyjne; aktywacja PPAR-γ, TRPV1, CB1R, DNA; ochrona przed stresem oksydacyjnym, podwyższony poziom endokannabinoidów; efekt chemoprewencyjny w modelu AOM – zmniejszona liczba guzów, ACF, polipów; aktywowana kaspaza-3, stłumione białko fosfo-Akt | ( 102 ) |
| in vitro (HCT116 i DLD-1) | Proapoptotyczne; aktywacja Noxa, podwyższenie ROS, indukcja stresu ER | ( 103 ) | |
| in vivo (samce myszy BALB/c, ksenograft-CT26) | Przeciwangiogenny, antymetastatyczny; hamowanie VEGF | ( 104 ) | |
| THC | in vitro (SW480, HCT-15, HT29, Caco-2, HCT116, SW620) | Proapoptotyczne; Aktywacja CB1 i hamowanie kaskady PI3K-AKT, RAS-MAPK, BAD i aktywacji kaspazy-3 | ( 106 ) |
Powolny rozwój i zatwierdzanie nowych leków przeciwnowotworowych na CRC wynika z braku odpowiednich modeli przedklinicznych. Modele 2D in vitro umożliwiają przeprowadzanie badań przesiewowych o wysokiej przepustowości i są proste w obsłudze, ale pozwalają jedynie na badanie interakcji komórka-komórka lub komórka-matryca, a nie całego TME; dlatego nie są fizjologicznie istotne i nie są klinicznie predykcyjne. Z drugiej strony, modele zwierzęce in vivo pozwalają na badanie interakcji całego organizmu z odpowiednim TME i heterogenicznością wewnątrzguzową, ale te modele nie nadają się do badań przesiewowych na dużą skalę, są bardzo czasochłonne i nie są „ludzkie”. Tak więc zarówno modele in vitro , jak i in vivo stanowią cenne narzędzie do badania karcynogenezy jelita grubego ( 107 ). Jednak ze względu na wspomniane różnice korelacja między tymi modelami nie jest zbyt silna ( 108 ). Z drugiej strony badania kliniczne są złotym standardem testowania i zatwierdzania każdego potencjalnego leku.
Ważne jest, aby wspomnieć o jednym badaniu klinicznym, które zbadało największą liczbę pacjentów onkologicznych otrzymujących medyczną marihuanę w latach 2015-2017 w Izraelu. Dwa tysiące dziewięćset siedemdziesięciu pacjentów cierpiących na raka piersi (20,7%), płuc (13,6%), trzustki (8,1%) i jelita grubego (7,9%) otrzymywało medyczną marihuanę jako leczenie paliatywne w celu złagodzenia objawów, takich jak ból, brak apetytu, złe samopoczucie, zaburzenia snu i nudności. W tym badaniu wykorzystano cztery rodzaje marihuany: szczepy sativa o wysokiej zawartości THC, bez CBD; szczepy indica o wysokiej zawartości THC bez CBD; szczep z równą ilością CBD i THC oraz szczepy bogate w CBD. Co ciekawe, większość pacjentów otrzymywała więcej niż jeden szczep. Dziewięćset dwóch (24,9%) pacjentów zmarło, a 682 (18,8%) pacjentów przerwało leczenie po 6 miesiącach obserwacji. Spośród pozostałych pacjentów 60,6% odpowiedziało na leczenie; U 95,9% pacjentów nastąpiła znacząca poprawa stanu, u 3,7% — brak zmian, u 0,3% — pogorszenie. Przed rozpoczęciem leczenia tylko 18,7% pacjentów stwierdziło dobrą jakość życia, natomiast po 6 miesiącach leczenia —69,5%. Spośród wszystkich objawów związanych z rakiem najbardziej poprawiły się nudności, wymioty, depresja, migrena i zaburzenia snu. Najczęstszymi skutkami ubocznymi leczenia konopiami po 6 miesiącach obserwacji były zawroty głowy, suchość w jamie ustnej i zwiększony apetyt. Psychoaktywne działania niepożądane zauważyło tylko 2,8% pacjentów. Co ciekawe, spośród 344 pacjentów przyjmujących opioidy, 36% z nich przerwało ich przyjmowanie. Stwierdzono, że medyczna marihuana jest dobrze tolerowaną i bezpieczną opcją terapii paliatywnej dla pacjentów onkologicznych ( 109 ).
Rola konopi w wrodzonej i adaptacyjnej odpowiedzi immunologicznej
Będąc środkami immunomodulującymi, ekstrakty z konopi i pojedyncze kannabinoidy mogą wpływać zarówno na wrodzoną, jak i adaptacyjną odpowiedź immunologiczną. Ogólnie rzecz biorąc, kannabinoidy są uważane za związki immunosupresyjne. Wpływają na wrodzoną odpowiedź immunologiczną poprzez hamowanie aktywności komórek NK, komórek dendrytycznych, migrację neutrofili i makrofagów z ich procesami prezentacji antygenu i fagocytozy ( 110 ) oraz poprzez wyzwalanie indukcji MDSC ( 111 , 112 ). Zapalenie jest głównym mechanizmem wrodzonej odpowiedzi immunologicznej. Ogólnie rzecz biorąc, kannabinoidy, takie jak THC i CBD, powodują zmniejszenie ekspresji cytokin prozapalnych i zwiększenie ekspresji cytokin przeciwzapalnych. Dzięki temu aktywnie hamują proces zapalny ( 57 ). Jednak niektóre badania wykazują, że związki te mają różny wpływ na stan zapalny, albo go wzmacniając, albo tłumiąc. Na przykład CBD może aktywować odpowiedź immunologiczną poprzez podniesienie ekspresji mRNA TNF-α, IL-6, jak wykazano u myszy w odpowiedzi na zapalenie płuc wywołane LPS ( 113 ). Natomiast CBD hamowało IL-6 i IL-8 w mysim modelu raka jelita grubego in vivo opartym na linii komórkowej CT26 ( 104 ). Te sprzeczne wyniki mogą być specyficzne dla tkanki i dawki.
Kanabinoidy mogą wpływać na adaptacyjne odpowiedzi immunologiczne poprzez oddziaływanie na odporność humoralną i komórkową. Odporność komórek T może być pod wpływem kanabinoidów na różne sposoby: mogą one wpływać na proliferację i liczbę komórek T poprzez polaryzację odpowiedzi cytokinowej na Th1 lub Th2 ( 114 ). Wykazano, że kanabinoidy hamują proliferację komórek T, powodują ich apoptozę i wspierają polaryzację Th2 ( 115 , 116 ). Niektóre wstępne badania in vitro i in vivo THC wykazały immunosupresyjny wpływ na komórki T i komórki B przy stosowaniu wysokich stężeń, podczas gdy efekty immunostymulujące obserwowano przy niskich stężeniach ( 110 ). Badania eksperymentalne przeprowadzone in vivo na makakach zakażonych wirusem SIV, które otrzymywały THC przez okres 17 miesięcy, wykazały wzrost liczby komórek T, zmniejszenie ładunku wirusowego i wzrost ekspresji cytokin Th2 ( 117 ). Inne badanie przeprowadzone na pacjentach z HIV wykazało wyższe stężenie komórek CD4+ i CD8+ u pacjentów z THC-dodatnim wynikiem w porównaniu z pacjentami z THC-ujemnym wynikiem ( 118 ). Jeśli chodzi o rolę CBD, wykazano również, że może ono działać jako immunosupresant Th2 in vitro i in vivo , polaryzując odpowiedź cytokinową na Th2 i działając jako immunostymulant na Th1 ( 119 ). Jeśli chodzi o odporność humoralną, niektóre raporty z badań na ludziach wykazały zmniejszoną liczbę limfocytów B i zmniejszoną ilość IgM i IgG po spożyciu kannabinoidów w postaci bhang ( 120 ).
Przyszłe perspektywy wzmocnienia immunoterapii za pomocą kannabinoidów i ekstraktów z Cannabis Sativa
Immunomodulacyjne działanie konopi jest dobrze udokumentowane. Obecnie istnieje wiele znanych odmian konopi, a każda z nich ma unikalny skład różnych związków. Wiele badań wykazało wpływ pojedynczych kannabinoidów, takich jak THC i CBD, na stany zapalne i wzrost komórek rakowych ( 98 ). Inne składniki rośliny (takie jak mniejsze kannabinoidy, terpeny, terpenoidy, flawonoidy i inne) mogą działać synergicznie z kannabinoidami i mogą być przydatne z terapeutycznego punktu widzenia. Modulujący wpływ tych związków jest znany jako „efekt otoczenia”; taka modulacja jest zazwyczaj dodatnia, co oznacza, że efekt leczniczy całego ekstraktu z rośliny jest bardziej znaczący niż efekt izolowanych związków ( 121 ). Podobnie jak w przypadku każdego innego leku, efekty w znacznym stopniu zależą od stężenia. W przyszłości, dzięki dalszym badaniom, możemy uzyskać więcej informacji na temat potencjalnego immunostymulującego wpływu pojedynczych kannabinoidów lub ekstraktów z konopi. Wiedza ta może pomóc personelowi medycznemu w integracji ekstraktów z konopi z ukierunkowaną terapią nowotworową, potencjalnie jako terapia wspomagająca. Należy zidentyfikować specjalne ekstrakty o silnym działaniu przeciwnowotworowym, które nie są cytotoksyczne dla normalnych komórek i mogą uwrażliwiać komórki nowotworowe na dalsze leczenie bez zmniejszania odpowiedzi immunologicznej. Następnie te ekstrakty można połączyć z immunoterapią, a takie połączenie może mieć działanie synergistyczne. Wyniki retrospektywnej analizy przeprowadzonej u pacjentów z czerniakiem, rakiem nerki i niedrobnokomórkowym rakiem płuc, gdy konopie były stosowane w połączeniu z lekiem immunoterapeutycznym Nivolumab, wykazały zmniejszenie RR, ale nie zmiany w PFS i OS. Potrzebne są dalsze badania w celu zbadania możliwych interakcji między kannabinoidami a lekami immunoterapeutycznymi ( 122 ).
Należy przeprowadzić dogłębną eksplorację badań nad konopiami i powiązanymi lekami. Obecnie dysponujemy ograniczonymi danymi na temat interakcji konopi z innymi lekami, zwłaszcza w przypadku terapii ukierunkowanej. Ponieważ inhibitory punktów kontrolnych układu odpornościowego są rodzajem najbardziej skutecznej i efektywnej immunoterapii dla pacjentów z rakiem jelita grubego. Dlatego też należy przeprowadzić badania nad możliwością wzmocnienia immunoterapii za pomocą ekstraktów z konopi ( Rysunki 2 , 3 ).
Rysunek 2.

Potencjał kannabinoidów w immunoterapii nowotworów. Górny panel pokazuje, jak kannabinoidy mogą zwiększyć immunogenność guza. Uwalnianie antygenów nowotworowych może być zwiększone ze względu na bezpośrednią cytotoksyczność kannabinoidów w komórkach nowotworowych. Następnie następuje prezentacja wzmocnionych antygenów nowotworowych, po której następuje wzrost odpowiedzi immunologicznej zależnej od limfocytów T i liza komórek nowotworowych przez limfocyty T. Dolny panel pokazuje, jak kannabinoidy mogą odwrócić immunosupresję guza. Makrofagi można przeprogramować do fenotypu przeciwnowotworowego za pomocą kannabinoidów. Immunostymulujące makrofagi M1 wydzielają cytokiny przeciwnowotworowe i skutecznie fagocytują komórki nowotworowe.
Rysunek 3.
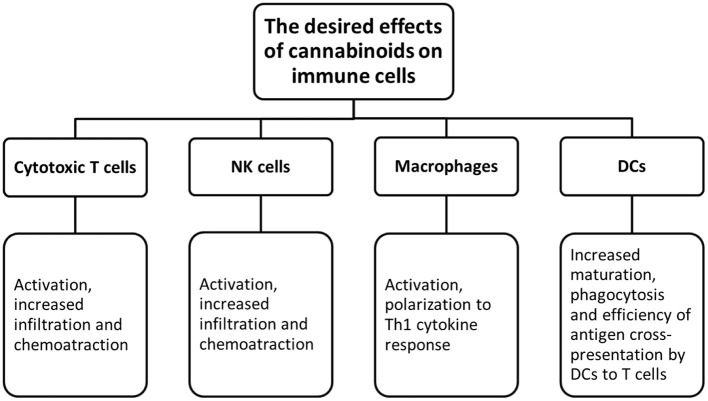
Pożądane efekty kannabinoidów na komórki odpornościowe.
Pierwsze podejście mogłoby się skupić na znalezieniu ekstraktów, które mogą zwiększyć immunogenność guza. Z tego powodu komórki nowotworowe będą bardziej podatne na rozpoznanie przez układ odpornościowy. W takim przypadku badania in vivo byłyby korzystne, aby zbadać konopie indyjskie jako terapię neoadjuwantową przed rozpoczęciem stosowania leków biologicznych. Powinny być one następnie poddane badaniom sprawdzającym, czy występuje wzmocnienie adaptacyjnych odpowiedzi immunologicznych pośredniczonych przez limfocyty T. Poprzez kierowanie cytotoksyczności do komórek rakowych, ekstrakty z konopi indyjskich mogą zwiększyć uwalnianie antygenów nowotworowych, a następnie wzmocnić prezentację antygenu. Należy również ocenić zmiany w mikrośrodowisku guza, takie jak naciek MDSC i Treg. Drugie podejście mogłoby się skupić na możliwości odwrócenia immunosupresji wywołanej przez guz za pomocą ekstraktów. Na przykład można to zrobić, przeprogramowując makrofagi na fenotyp przeciwnowotworowy. Będąc wysoce plastycznymi komórkami, makrofagi mogą łatwo przejść z typu pro-nowotworowego na typ przeciwnowotworowy. Niektóre badania in vivo wykazały, że wpływanie na szlak PI3kγ w makrofagach może dalej prowadzić do ich polaryzacji do typu immunostymulującego poprzez zwiększenie cytotoksyczności komórek CD8+ T i poprawę odpowiedzi na ICIs ( 123 ). Znajdując ekstrakty, które mogą wpływać na ten konkretny szlak, można połączyć je z immunoterapią, aby wykazać działanie synergistyczne. Ponadto ocenę polaryzacji makrofagów można przeprowadzić poprzez badanie cytokin. Ekstrakty z konopi, które polaryzują makrofagi do typu M1 i nie posiadają właściwości przeciwzapalnych, można dalej łączyć z immunoterapią.
Wniosek
W literaturze silnie sugerowano, że kannabinoidy i ekstrakty z konopi mogą być stosowane w leczeniu raka jelita grubego. Dowody wskazują, że kannabinoidy mają duży potencjał przekształcenia się w obiecujące leki. Oczywiste jest, że związki te mogą oddziaływać na kluczowe szlaki sygnałowe rozwoju raka. Ponadto należy przeprowadzić więcej przedklinicznych i klinicznych ocen kannabinoidów jako środków przeciw CRC i immunomodulujących. Dodatkowe badania pomogłyby nam znaleźć nowe możliwości zapobiegawcze i terapeutyczne dla pacjentów, u których istnieje ryzyko rozwoju CRC lub którzy obecnie się z nim zmagają. Wprowadzając te silne związki do obecnych protokołów leczenia, można osiągnąć redukcję dawki innych leków, które są wysoce toksyczne, zmniejszając w ten sposób niepożądane skutki uboczne; podobnie kannabinoidy mogą prawdopodobnie uwrażliwić komórki złośliwe na dalszą ukierunkowaną terapię.
Ponadto dalsze badania nowotworów, zwłaszcza ich sekwencjonowanie, dostarczą więcej informacji na temat ich specyficznych cech. Wiedza o tym, w jaki sposób ścieżki genetyczne oddziałują z pewnymi kannabinoidami, może pomóc lekarzom przepisywać je zgodnie z genetycznym składem nowotworu. Z tego powodu personel medyczny będzie przepisywał terapie oparte na kannabinoidach konkretnemu pacjentowi z konkretną chorobą nowotworową. Leczenie stanie się zorientowane na pacjenta i specyficzne dla wskazań. W rezultacie rokowanie w przypadku nowotworu i jego wskaźnik przeżycia mogą się znacznie poprawić.
źródło: https://pmc.ncbi.nlm.nih.gov/articles/PMC8497796/