W ciągu ostatnich kilku lat byliśmy świadkami wzrostu liczby nowych doniesień dotyczących roli kannabinoidów, zarówno syntetycznych, jak i ziołowych, w mechanizmach zapalenia i karcynogenezy. Jednakże pomimo bogactwa danych in vitro i niepotwierdzonych doniesień, brakuje dowodów na to, że kannabinoidy mogą działać jako korzystne leki w leczeniu nieswoistego zapalenia jelit (IBD) lub rozwoju nowotworu przewodu pokarmowego u człowieka. Pewien wgląd w wpływ marihuany medycznej (zwykle oznaczającej suszone kwiaty) i kannabinoidów na IBD uzyskano dzięki kwestionariuszom i małym badaniom pilotażowym. Jeśli chodzi o raka jelita grubego, dostępne są jedynie dane przedkliniczne. Obecnie Δ<sup>9</sup>-tetrahydrokannabinol (THC) i jego syntetyczne formy, dronabinol i nabilon, stosuje się jako leczenie uzupełniające w celu łagodzenia przewlekłego bólu i spastyczności u pacjentów ze stwardnieniem rozsianym, a także nudności wywołanych chemioterapią . Używanie marihuany medycznej jest dozwolone tylko w ograniczonej liczbie krajów. Żadna z wymienionych substancji nie jest obecnie wskazana w leczeniu IBD. Niniejsza recenzja stanowi aktualizację naszej wiedzy na temat roli kannabinoidów w zapaleniu jelit i karcynogenezie oraz dyskusję na temat ich potencjalnego zastosowania terapeutycznego.
Korzystne właściwości kannabinoidów w przewodzie żołądkowo-jelitowym (GI) wynikają z faktu, że jelita są wyposażone w układ endokannabinoidowy (ECS), sieć regulacyjną receptorów kannabinoidowych, enzymów i ligandów, które odgrywają kluczową rolę w procesach fizjologicznych, jak i fizjologicznych. procesy patofizjologiczne [ 1 , 2 , 3 , 4 ]. Tak więc w jelitach występuje ekspresja klasycznych receptorów kannabinoidowych 1 i 2 (CB 1 , CB 2 ) oraz niereagujących na kannabinoidy receptorów CB 1 /CB 2 (receptor sprzężony z białkiem G 55 [GPR55], członek podrodziny kanałów kationowych o potencjale przejściowym 1 [TRPV1] oraz receptory gamma i alfa aktywowane przez proliferatory peroksysomów [PPARγ i PPARα]). Ponadto enzymy wytwarzające endokannabinoidy (EC), takie jak specyficzna dla N-acylofosfatydyloetanoloaminy fosfolipaza D i lipaza diacyloglicerolu, oraz enzymy rozkładające EC, takie jak hydrolaza amidów kwasów tłuszczowych (FAAH) i lipaza monoacyloglicerolu (MGL), znajdują się w jelito. CB 1 i CB 2 lokalizują się głównie w nerwach jelitowych, nabłonku jelitowym i komórkach odpornościowych o zmiennej ekspresji [ 5 , 6 , 7 ]. Podczas gdy CB 1 ulega ekspresji na wysokim poziomie w cholinergicznych neuronach jelitowych [ 8 ], CB 2 ulega dużej ekspresji w komórkach odpornościowych [ 9 ].
Anandamid (AEA) i 2-arachidonoiloglicerol (2-AG) to najlepiej opisane EC, które aktywują receptory CB 1 , CB 2 i wyżej wymienione receptory inne niż CB 1 /CB 2 [ 10 , 11 , 12 ]. Oprócz tych składników ECS w przewodzie pokarmowym znaleziono lipidy podobne do EC, głównie N-acyloetanoloaminy, takie jak palmitoiloetanoloamid (PEA) i oleiloetanoloamid. Tam działają na receptory inne niż CB 1 /CB 2 , takie jak GPR55 i GPR119 i wpływają na sygnalizację AEA, zwaną także „efektem otoczenia” [ 13 ]. EC są najprawdopodobniej zaangażowane w liczne mechanizmy regulacyjne, np. utrzymują nienaruszoną barierę nabłonkową [ 14 , 15 ] i utrzymują tolerancję immunologiczną poprzez kontrolowanie ekspansji regulatorowego podzbioru limfocytów T Tr1 i obecność immunosupresyjnych makrofagów CX3CR1hi [ 16 ] . Nieoczekiwanie donoszono o zmianach w poziomach EC i lipidów podobnych do EC u pacjentów z nieswoistym zapaleniem jelit (IBD) [ 17 ] i rakiem jelita grubego (CRC) [ 18 ]. Nie wiadomo jednak, czy zmiany te rzeczywiście korelują z postępem choroby. Jeśli chodzi o CRC, dane in vitro z linii komórkowych raka okrężnicy przekonująco pokazują antyproliferacyjne działanie kannabinoidów [ 19 ] i w rzeczywistości modele CRC u myszy z nokautem sugerują antyonkogenną rolę co najmniej CB 1 [ 20 , 21 ]. Jednakże w odniesieniu do CB 2 badania na ludzkich pacjentach z CRC pokazują, że jego ekspresja koreluje ze zmniejszonym przeżyciem [ 22 ]. W podobny sposób wykazano, że ekspresja CB 1 koreluje z gorszym współczynnikiem przeżycia w stabilnym mikrosatelitarnym CRC w stadium II [ 23 ]. Dlatego doniesienia z badań na ludziach i eksperymentów są kontrowersyjne co do korzystnej roli ECS w CRC. W poniższych rozdziałach pokrótce podsumowano najnowsze wyniki dotyczące roli ECS i działania kannabinoidów w IBD i CRC, po czym nastąpiła dyskusja na temat potencjalnej terapii kannabinoidami.
Kannabinoidy i IBD
IBD, którego głównymi objawami są wrzodziejące zapalenie jelita grubego i choroba Leśniowskiego-Crohna (CD), charakteryzuje się przewlekłymi i nawracającymi atakami zapalnymi przewodu pokarmowego [ 24 ]. Chociaż szczegółowe mechanizmy są nadal nieznane, niekontrolowana i źle ukierunkowana odpowiedź immunologiczna przeciwko antygenom drobnoustrojów w połączeniu z predyspozycjami genetycznymi i czynnikami środowiskowymi przyczynia się do wieloczynnikowego pojawienia się choroby [ 24 , 25 ].
Spostrzeżenia z modeli zwierzęcych
Dane funkcjonalne służące do badania roli receptorów kannabinoidowych i ECS w IBD uzyskano głównie z modeli zwierzęcych i wykazały one, że składniki ECS ulegają zmianie podczas eksperymentalnego zapalenia jelit. Zatem doniesienia o podwyższonych poziomach CB 1 , CB 2 i AEA sugerują zwiększoną sygnalizację kannabinoidową w stanach zapalnych [ 26 , 27 , 28 ]. Farmakologiczna aktywacja CB 1 i CB 2 łagodzi eksperymentalne zapalenie jelita grubego [ 28 , 29 , 30 ], natomiast stosowanie antagonistów lub ablacja genetyczna receptorów kannabinoidowych nasila stan zapalny [ 26 , 31 ]. Co więcej, genetyczny niedobór FAAH lub hamowanie FAAH lub MGL ( a w konsekwencji wzrost AEA lub 2-AG) okazały się chronić przed zapaleniem okrężnicy wywołanym przez siarczan dekstranu sodu (DSS) lub kwas trinitrobenzenosulfonowy (TNBS) [ 31,32 , 33 , 34 , 35 , 36 ].
Kilka fitokannabinoidów, a także syntetycznych analogów i związków podobnych do EC wykazało korzystne działanie w zwierzęcych modelach zapalenia jelit. Psychotropowe Δ 9 -tetrahydrokannabinol (THC) i niepsychotropowy kannabidiol (CBD) zmniejszały zapalenie jelita grubego wywołane TNBS w okrężnicy szczura [ 37 ]. W przeciwieństwie do THC, CBD ma wyjątkowo niskie powinowactwo do CB 1 i CB 2 , ale wykazano, że ma antagonistyczne działanie na GPR55 [ 11 ]; Opisano, że CBD działa na PPARγ [ 38 ] i TRPV1 oraz hamuje aktywność FAAH, zmieniając w ten sposób poziomy EC [ 39 , 40 ]. Wykazano także, że wywiera działanie ochronne w modelu zapalenia okrężnicy wywołanego kwasem dinitrobenzenosulfonowym (DNBS) u myszy [ 41 ]. Nowsze prace wykazały, że ekstrakt z Cannabis sativa o wysokiej zawartości CBD, a nie samo CBD, zapewnia ochronę [ 34 ]. Wykazano , że analog CBD O-1602, który również nie ma powinowactwa do CB 1 i CB 2 , ale ma właściwości agonistyczne w stosunku do GPR55 [ 11 ], ma działanie przeciwzapalne w zapaleniu jelita grubego; jednakże nie pośredniczył w nich GPR55 [ 42 ]. Wydaje się, że O-1602 pośredniczy w zmniejszaniu motoryki okrężnicy poprzez GPR55 [ 43 ]. Prace w naszym własnym laboratorium wykazały, że GPR55 może mieć działanie prozapalne, ponieważ antagonista GPR55 i genetyczna delecja genu GPR55 złagodziły zapalenie jelita grubego DSS u myszy [ 44 ]. Wykazano, że kannabigerol, niepsychotropowy fitokannabinoid, łagodzi zapalenie okrężnicy u myszy i zmniejsza wytwarzanie tlenku azotu w makrofagach oraz powstawanie reaktywnych form tlenu w komórkach nabłonka jelitowego [ 45 ]. Innym przeciwzapalnym i niepsychotropowym fitokannabinoidem jest kannabichromen, który, jak wykazano, hamuje inaktywację EC [ 46 ].
Syntetyczny, nieselektywny agonista CB 1 /CB 2 WIN 55,212-2 został niedawno przetestowany w leczeniu zapalenia jelita grubego wywołanego DSS i wykazał właściwości ochronne i przeciwzapalne, które wydają się być przynajmniej częściowo pośredniczone przez hamowanie kinazy białkowej aktywowanej mitogenem p38 [ 47 ] . Innym nieselektywnym agonistą receptora kannabinoidowego ostatnio testowanym w leczeniu zapalenia jelita grubego wywołanego DSS jest HU210 [ 48 ]. Wcześniej wykazano, że ma działanie ochronne w modelu zapalenia okrężnicy wywołanego DNBS [ 26 ], substancja ta utrzymuje integralność funkcji bariery jelitowej niezależnie od receptora Toll-podobnego 4, ale wywołuje pozajelitowe działanie przeciwzapalne zależne od receptora Toll-podobnego 4- za pośrednictwem aktywacji kinazy białkowej p38 aktywowanej mitogenem [ 48 ].
Związek podobny do EC, PEA, jest wytwarzany endogennie w wyniku urazu zapalnego i opisano, że działa poprzez CB1 , CB2 , GPR55, PPARα i TRPV1 [ 49 ]. W kilku modelach chemicznie wywołanego zapalenia okrężnicy u myszy wykazano, że PEA zmniejsza stan zapalny [ 49 , 50 ] i chroni przepuszczalność jelit przez PPARα, CB2 i GPR55 [ 49 ]. Zgodnie z tymi wynikami, hamowanie enzymu degradującego PEA, N-acyloetanoloaminy, hydrolizującej kwaśnej amidazy, zwiększało poziomy PEA, a także chroniło przed zapaleniem jelita grubego [ 51 ]. Ponadto Sarnelli i in. [ 52 ] podali, że hamowanie angiogenezy związanej ze stanem zapalnym przez PEA w modelu DSS było zależne od PPARα. Dodatkowo wykazali, że uwalnianie czynnika wzrostu śródbłonka naczyniowego i tworzenie naczyń uległy zmniejszeniu, co doprowadziło do zmniejszenia uszkodzenia błony śluzowej [ 52 ]. Niedawno odkryto, że analog PEA, adelmidrol, wywiera działanie przeciwzapalne, w którym częściowo pośredniczy PPARγ [ 53 ].
Podsumowując, chociaż nadal brakuje szczegółowych mechanizmów działania wszystkich tych związków, dane przedkliniczne są obiecujące i sugerują, że syntetyczne i ziołowe kannabinoidy, inhibitory FAAH i MGL (a w konsekwencji zwiększone poziomy EC i lipidów EC) mogą być przydatne w leczeniu leczyć IBD. Niemniej jednak znalezienie ukierunkowanego leczenia, szczególnie do długotrwałego stosowania i wywołującego niewielkie skutki uboczne, będzie trudne, biorąc pod uwagę, że ECS jest obecny w różnych tkankach całego organizmu i bierze udział w wielu procesach fizjologicznych.
Kannabinoidy i CRC
Obserwacje zmienionych poziomów ekspresji składników ECS w biopsjach nowotworów wskazują na kluczową rolę ECS w rozwoju CRC. W zmianach CRC zwiększył się poziom AEA i 2-AG oraz enzymów odpowiedzialnych za syntezę i degradację EC, co wskazuje na zwiększony metabolizm EC [ 18 , 54 , 55 ]. Jednakże, w porównaniu z sąsiadującą nienowotworową błoną śluzową okrężnicy, stwierdzono, że ekspresja CB 1 jest obniżona w próbkach CRC w wyniku hipermetylacji DNA wysp CpG w regionie promotora CNR1 (gen kodujący CB 1 ) [ 20 , 21 , 56 ] . W ciągu ostatnich lat opublikowano mnóstwo badań przedklinicznych, w których opisano możliwe właściwości przeciwnowotworowe (endo)kannabinoidów, w tym właściwości antyproliferacyjne, proapoptotyczne, antyangiogenne, przeciwmigracyjne i przeciwinwazyjne. Mechanizmy molekularne, dzięki którym kannabinoidy wywierają działanie przeciwnowotworowe, zostały bardzo szczegółowo podsumowane w innym miejscu [ 57 , 58 , 59 , 60 ]. Chociaż dane z testów komórkowych wydają się bardzo obiecujące, dane uzyskane z modeli in vivo są skąpe i tworzą dość skomplikowany obraz. Na przykład leczenie nieselektywnym agonistą CB1 / CB2 HU210 lub CBD zmniejszyło rozwój zmian przednowotworowych w mysich modelach chemicznie wywołanego CRC [ 61 , 62 ]. Podobne wyniki uzyskano zwiększając poziom EC poprzez hamowanie enzymów degradujących FAAH i MGL [ 61 , 63 ]. Jednakże inne badanie wykazało, że blokada sygnalizacji ECS przez zastosowanie antagonisty CB1 SR141716 również zmniejszyła powstawanie zmian przednowotworowych w mysim modelu CRC [ 64 ].
Ostatnio wyszło na jaw, że GPR55, nietypowy receptor kannabinoidowy reagujący na niektóre (endo)kannabinoidy, wywiera działanie sprzyjające rozwojowi nowotworu poprzez zwiększenie masy guza i przerzutów u myszy [ 21 , 65 ]. Podkreśla to pogląd, że ECS i powiązane z nim składniki pełnią różnorodne i często przeciwstawne funkcje oraz że niezbędne jest lepsze zrozumienie podstawowych mechanizmów i interakcji receptorów. Na przykład konieczne będzie uzyskanie większej ilości informacji na temat ich wpływu na populację komórek odpornościowych związaną z nowotworami, tj. mikrośrodowisko nowotworu, zanim kannabinoidy będą mogły zostać przeniesione do kliniki. Ponieważ immunoterapię nowotworową wprowadza się obecnie nawet w przypadku guzów litych [ 66 ], dość niepokojące jest to, że jak dotąd tylko kilka badań dotyczyło wpływu kannabinoidów na mikrośrodowisko guza. Doniesiono, że WIN 55,212-2 (nieselektywny agonista CB1 / CB2 ) i JWH133 (selektywny agonista CB2 ) mają wyraźniejsze działanie przeciwnowotworowe na ksenoprzeszczepy ludzkiego czerniaka u myszy z prawidłową odpornością niż na te wszczepione myszom SCID [ 67 ], co wskazuje, że działanie przeciwnowotworowe (endo)kannabinoidów odbywa się częściowo za pośrednictwem komórek odpornościowych. Jednak inne badanie wykazało, że THC hamuje reaktywność immunologiczną gospodarza przeciwko rakowi płuc u myszy, promując w ten sposób wzrost nowotworu [ 68 ]. To odkrycie zostało potwierdzone przez Hegde i in. [ 69 ], którzy podali, że podawanie THC spowodowało masową ekspansję komórek supresorowych pochodzących z szpiku, heterogennej populacji komórek o silnych właściwościach immunosupresyjnych. Dodatkowo niedawno wykazaliśmy, że skład komórek odpornościowych obecnych w nowotworach jelita grubego u myszy z niedoborem GPR55 był zmieniony w porównaniu z myszami typu dzikiego [ 21 ].
Podsumowując, wydaje się, że wpływ (endo)kannabinoidów na nowotwory jest znacznie bardziej zróżnicowany, niż obecnie szacuje się na podstawie obiecujących danych in vitro. Dlatego możemy potrzebować bardziej dokładnych badań podstawowych na temat interakcji ECS z przedziałem odpornościowym nowotworu przed rozpoczęciem badań klinicznych z kannabinoidami lub medyczną marihuaną jako metodą leczenia przeciwnowotworowego.
Czy kannabinoidy są opcją terapeutyczną w przypadku zapalenia i nowotworu przewodu pokarmowego?
Konopie indyjskie /kannabinoidy w IBD
Zapalenie jelit jest wysoce podatne na leczenie kannabinoidami, co udokumentowano w przypadku IBD u ludzi nie tylko niepotwierdzonymi doniesieniami, ale, co ważne , także kilkoma badaniami kwestionariuszowymi [ 70,71,72,73 ] , a także badaniami obserwacyjnymi i prospektywnymi badaniami klinicznymi [ 70,71,72,73 ] , 74 , 75 , 76 , 77 ]. Wiele kwestionariuszy ujawniło, że pacjenci z IBD często samoleczą się konopiami indyjskimi, aby złagodzić ból brzucha i biegunkę. Jednak długotrwałe używanie konopi indyjskich nie zawsze może być korzystne. Ankieta przeprowadzona przez Storr i in. [ 72 ] wykazali, że używanie konopi indyjskich przez ponad 6 miesięcy było silnym czynnikiem predykcyjnym konieczności przeprowadzenia operacji u pacjentów z CD. Badania prospektywne również wykazały mieszane wyniki dotyczące skuteczności leczenia kannabinoidami u pacjentów z IBD. Podczas gdy małe badanie z udziałem 21 pacjentów z CD wykazało, że krótkotrwałe stosowanie (8 tygodni) konopi indyjskich bogatych w THC spowodowało obniżenie wskaźników aktywności choroby Leśniowskiego-Crohna u prawie wszystkich pacjentów [ 76 ], inne badanie wykazało, że 2-miesięczne leczenie w umiarkowanej CD z niepsychoaktywnym CBD nie było korzystne [ 78 ]. Dodatkowo przeprowadzono eksperymenty na ludzkich tkankach okrężnicy, które dostarczyły informacji na temat przydatności terapii IBD opartej na kannabinoidach. Wykorzystując próbki tkanek okrężnicy od pacjentów z IBD i wyrostków robaczkowych, Couch i in. [ 79 ] wykazali, że inkubacja z CBD i PEA zapobiegała wytwarzaniu cytokin zapalnych odpowiednio poprzez szlaki CB2 i TRPV1 oraz poprzez PPARα. Inne badanie z użyciem eksplantatów od pacjentów z IBD wykazało, że ekstrakt z konopi indyjskich tłumił ekspresję cyklooksygenazy-2 i metaloproteinazy-9 [ 80 ]. Autorzy opisali kwas Δ 9 -tetrahydrokannabinolowy jako aktywny, niepsychotropowy składnik przeciwzapalny ekstraktu, który może być przydatny w leczeniu IBD zamiast CBD.
Podsumowując, badania te sugerują, że leczenie konopiami indyjskimi i kannabinoidami rzeczywiście może złagodzić stan zapalny w IBD, ale najprawdopodobniej zależy to od dawki, sposobu stosowania, a w konsekwencji stężenia w tkance, które można osiągnąć za pomocą kannabinoidów. Jednak wadą jest to, że leczenie kannabinoidami aktywującymi receptor CB 1 wiąże się z psychotropowymi skutkami ubocznymi. THC i jego pochodne mogą powodować zawroty głowy, senność, euforię i halucynacje w wyniku aktywacji CB 1 w mózgu [ 81 ]. Problem ten można obejść, stosując kannabinoidy działające obwodowo, co wydaje się wysoce prawdopodobne, biorąc pod uwagę znaczną obecność ECS w jelitach. Jednakże na motorykę przewodu pokarmowego duży wpływ ma ośrodkowa CB 1 [ 82 ], a obwodowo ograniczeni agoniści CB 1 /CB 2 podani dootrzewnowo nie poprawili stanu zapalnego w eksperymentalnych modelach zapalenia jelit [ 83 , 84 ]. Zamiast tego działały ochronnie, gdy były stosowane do komory mózgowej [ 84 ]. Ponadto niedobór CB 1 w nerwie błędnym spowalnia motorykę przewodu pokarmowego [ 85 ]. Obserwacje te wskazują, że obwodowa aktywacja receptorów kannabinoidowych może nie być wystarczająca do złagodzenia stanu zapalnego przewodu pokarmowego i że centralna aktywacja receptorów kannabinoidowych jest konieczna do pełnego procesu gojenia. Oś jelitowo-mózgowa również może odgrywać ważną rolę w tym procesie.
Inną opcją minimalizacji niepożądanych skutków psychotropowych podczas leczenia zapalenia przewodu pokarmowego może być wybór niższej dawki kannabinoidów. Z badań przedklinicznych wiadomo, że stosowanie niskich dawek THC poprawia działanie np. na miażdżycę [ 86 ] i funkcje poznawcze [ 87 ]. U pacjentów z przewlekłym zespołem górnego neuronu ruchowego leczenie nabilonem w dawce 1 mg/dzień znacząco zmniejszyło ból [ 88 ]. Co ważne, w tym badaniu nabilon był dobrze tolerowany. Badanie kliniczne przeprowadzone na starszych pacjentach z demencją wykazało również, że niskie dawki THC (0,75 mg dwa razy dziennie) są bezpieczne i dobrze tolerowane; nie było różnic w odczuwaniu „haju” pomiędzy grupą otrzymującą lek i grupą placebo [ 89 ]. Ze względu na znaczącą ekspresję CB 1 w mózgu, jelitowym układzie nerwowym i nabłonku jelit, leczenie małymi dawkami kannabinoidów w IBD może aktywować receptory kannabinoidowe zarówno ośrodkowo, jak i obwodowo, przyczyniając się w ten sposób do gojenia się ran, przywrócenia funkcji barierowych i rozluźnienia jelit.
Wreszcie, inhibitory enzymów degradujących EC podnoszą poziomy EC i są odpowiednie do leczenia zapalenia przewodu pokarmowego. Na podstawie wyników badań przedklinicznych można stwierdzić, że inhibitory MGL mogą być obiecującymi kandydatami do leczenia IBD [ 32 ], ale również inhibitory FAAH okazały się skuteczne w łagodzeniu eksperymentalnego zapalenia jelit [ 35 ]. Jednakże przełożenie tych skutków na choroby przewodu pokarmowego u ludzi jest opóźnione, szczególnie w przypadku inhibitorów FAAH, ponieważ badania kliniczne zostały wstrzymane ze względu na incydent z BIA 10-2474 [ 90 ]. Ponadto możliwość wystąpienia sercowo-naczyniowych skutków ubocznych inhibitorów FAAH może również ograniczać ich stosowanie u ludzi [ 91 , 92 , 93 ].
Konopie indyjskie /kannabinoidy w CRC
Korzystne działanie kannabinoidów na CRC znane jest jedynie z eksperymentów na myszach. Nokaut CB 1 u myszy Apc Min/+ i C57BL/6 [ 20 , 21 ] powoduje, w zależności od zastosowanego modelu, zwiększone obciążenie nowotworem w jelicie cienkim i okrężnicy, co wskazuje na supresorową rolę CB 1 w CRC. Natomiast CB 2 powiązano z postępem CRC [ 22 , 94 ], co utrudnia ustalenie jasnego uzasadnienia leczenia kannabinoidami u ludzi CRC. Niezbędnych jest zatem więcej danych przedklinicznych na temat roli CB 2 w doświadczalnym CRC, aby stworzyć podstawę do przełożenia wyników eksperymentalnych na praktykę kliniczną. Ponieważ kannabinoidy zostały zatwierdzone do leczenia wymiotów i nudności u pacjentów chorych na nowotwory poddawanych chemioterapii, z obserwacji tych pacjentów możemy dowiedzieć się, czy przeciwwymiotne leczenie kannabinoidami wpływa na progresję nowotworu podczas chemioterapii. W tym kontekście należy wspomnieć, że połączenie THC z CBD skutecznie zwiększa śmierć komórek i migrację linii komórkowych szpiczaka mnogiego w synergii z karfilzomibem, inhibitorem proteasomu, stosowanym w leczeniu szpiczaka mnogiego [ 95 ]. Temat terapii kannabinoidami w CRC został również poruszony w niedawnym artykule redakcyjnym [ 63 ].
Wniosek
Eksperymentalne modele zapalenia jelit pokazują, że pomocne może być leczenie kannabinoidami. Również w modelach eksperymentalnych CRC wykazano , że CB 1 ma działanie ochronne. Jednakże jeśli chodzi o mechanizmy działania, wiadomo, że kannabinoidy mogą przekazywać sygnał wieloma szlakami, najprawdopodobniej w sposób oparty na ligandach [ 96 ]. Pomimo przytłaczającej ilości danych wskazujących, że kannabinoidy mają właściwości przeciwnowotworowe in vitro, dane in vivo potwierdzające ich działanie są nieliczne. Niektóre receptory reagujące na kannabinoidy, takie jak GPR55, wykazały nawet właściwości prozapalne i prokarcynogenne. Zatem ze względu na złożoność mechanizmów w obrębie ECS, korzystny wpływ fitokannabinoidów i syntetycznych kannabinoidów na ludzi nie zawsze jest pewny. Ponadto psychotropowe działanie THC i syntetycznych agonistów CB 1 w dalszym ciągu utrudnia ich szersze zastosowanie, czego można uniknąć poprzez strategie takie jak stosowanie kannabinoidów w małych dawkach z minimalnymi efektami ośrodkowymi lub stosowanie ligandów receptorów kannabinoidowych działających obwodowo. Każde podejście ma wady i wiele pytań. Z pewnością potrzebne są duże badania kliniczne, aby stworzyć solidne wytyczne dotyczące bezpieczeństwa, skuteczności, dawkowania, drogi, formy stosowania i skutków ubocznych kannabinoidów. Mamy nadzieję, że w najbliższej przyszłości dane kliniczne zapewnią podstawę do tego, czy konopie indyjskie lub kannabinoidy mogą stać się wartościowymi lekami, przynajmniej w leczeniu IBD. W przypadku CRC zajmie to z pewnością więcej czasu.
Rak jelita grubego jest poważnym problemem zdrowia publicznego. Niestety, obecnie nie istnieje żadna skuteczna opcja leczenia tego typu nowotworu złośliwego. Najbardziej obiecującym obecnie leczeniem raka jest immunoterapia, która jest również nazywana terapią biologiczną lub ukierunkowaną. Ten rodzaj terapii wzmacnia zdolność układu odpornościowego pacjenta do zwalczania nowotworu złośliwego. Jednak komórki nowotworowe mogą stać się oporne na immunoterapię i uniknąć nadzoru immunologicznego poprzez uzyskanie zmian genetycznych. Dlatego też konieczne są nowe strategie leczenia. W ostatniej dekadzie kilka raportów sugeruje skuteczność kannabinoidów i ekstraktów z Cannabis sativa w hamowaniu proliferacji nowotworów in vitro i in vivo , w tym nowotworów złośliwych jelit. Wykazano, że kannabinoidy modulują ścieżki zaangażowane w proliferację komórek, angiogenezę, zaprogramowaną śmierć komórek i przerzuty. Z tego powodu są proponowane jako terapia wspomagająca w przypadku wielu nowotworów złośliwych. O wiele mniej informacji istnieje na temat potencjału stosowania konopi w połączeniu z immunoterapią. W tym artykule analizujemy możliwość wykorzystania kannabinoidów w modulacji immunoterapii raka jelita grubego i omawiamy możliwe zalety i ograniczenia.
Obecnie rak jelita grubego (CRC) jest uważany za trzeci najbardziej śmiertelny i czwarty najczęściej wykrywany nowotwór na świecie ( 1 ). Pomimo obecności wysoce zaawansowanych technik przesiewowych, wskaźnik zapadalności stale rośnie na całym świecie ( 2 ). Szacuje się, że globalne obciążenie rakiem jelita grubego wzrośnie o 60% do ponad 2,2 miliona nowo zdiagnozowanych przypadków i 1,1 miliona zgonów do 2030 roku ( 3 ). Czynniki takie jak siedzący tryb życia, zwiększone spożycie alkoholu, tytoniu, czerwonego mięsa, predyspozycje genetyczne, przewlekłe procesy zapalne przewodu pokarmowego są czynnikami wyzwalającymi tego typu złośliwość ( 4 ). Wiadomo, że głównymi prekursorami CRC są polipy gruczolakowate. Szybkość transformacji tych polipów w raka wynosi ~0,25% rocznie. Gdy te zmiany mają wysoki stopień dysplazji i architekturę kosmków, ryzyko transformacji w nowotwór złośliwy wzrasta do 50% ( 5 ).
Zrozumienie patogenezy raka jelita grubego jest bardzo ważne dla wyboru właściwej terapii. Etiologia CRC jest złożona i obejmuje akumulację nabytych modyfikacji epigenetycznych i genetycznych, które przekształcają normalne komórki nabłonkowe w złośliwe. Klasyczny model progresji nowotworu nazywa się rozwojem sekwencji polipa-raka, która obejmuje trzy główne etapy. Pierwszym etapem jest tworzenie łagodnych nowotworów, takich jak gruczolaki i siedzące ząbkowane polipy. Drugi etap charakteryzuje się progresją łagodnych nowotworów w nowotwory bardziej zaawansowane histologicznie, a ostatni etap – ich transformacją w raka. Proces ten może trwać wiele lat bez wykazywania jakichkolwiek oznak i objawów. Kiedy CRC się rozwinie, może minąć jeszcze kilka lat, zanim zostanie zdiagnozowany. CRC jest spowodowany mutacjami w onkogenach, genach supresorowych nowotworu i genach zaangażowanych w mechanizmy naprawy DNA. Jedna z pierwszych mutacji występuje zazwyczaj w genie polipowatości gruczolakowatej jelita grubego (APC), który jest supresorem nowotworu, po czym następują mutacje w genach KRAS, TGF-β, BAX, BRAF i innych ( 6 ).
Większość przypadków CRC jest sporadyczna (70–80%), podczas gdy dziedziczne i rodzinne przypadki CRC stanowią odpowiednio około 5 i 25%. Sporadyczne nowotwory powstają z powodu mutacji punktowych, a molekularna patogeneza tych nowotworów jest bardzo niejednorodna. Dziedziczna grupa tego konkretnego nowotworu złośliwego jest spowodowana odziedziczonymi mutacjami i może być podzielona na dwie grupy: polipowatość i niepolipowatość. Typ polipowatości obejmuje głównie rodzinną polipowatość gruczolakowatą, która charakteryzuje się obecnością licznych, prawdopodobnie złośliwych polipów w jelicie grubym. Wariant niepolipowatości jest reprezentowany przez zespół Lyncha ( 7 ). Rodzinny CRC jest również spowodowany odziedziczonymi mutacjami i występuje rodzinnie bez obecności konkretnych dziedzicznych zespołów ( 8 ).
Ostatnio zaproponowano dwie molekularne klasyfikacje patologiczne oparte na szeroko zakrojonej analizie genomicznej i transkryptomowej CRC. Pierwsza z nich nazywa się The Cancer Genome Atlas (TCGA) i obejmuje trzy grupy: hipermutacje (13%), ultramutacje (3%) i niestabilność chromosomową (84%). Kategoria hipermutacji charakteryzuje się wysokim wskaźnikiem mutacji, wadliwą naprawą niedopasowań (dMMR) z dobrym rokowaniem, ale złym rokowaniem po nawrocie. Typ ultramutacji ma niezwykle wysoki wskaźnik mutacji z mutacją korekty epsilon polimerazy DNA i ogólnie dobrym rokowaniem. Większość CRC wyróżnia się niestabilnością chromosomową (CIN) z cechami niskiej częstości mutacji, ale wysoką częstością zmian liczby kopii somatycznych DNA. Druga klasyfikacja oparta na ekspresji genów nazywa się Consensus Molecular Subtypes (CMS) i obejmuje cztery grupy. CMS1 (14%) charakteryzuje się niestabilnością mikrosatelitarną (MSI), mutacją onkogenu BRAF i silną aktywacją immunologiczną. U pacjentów z tym podtypem zauważono słaby wskaźnik przeżycia po nawrocie. CMS2 (37%), zwany również kanonicznym, wykazuje wysoką niestabilność chromosomową i aktywację sygnalizacji WNT i MYC. CMS3 (13%), znany jako metaboliczny, ma liczne mutacje KRAS i deregulowane ścieżki metabolizmu. CMS4 (23%), zwany mezenchymalnym, jest opisany obecnością nacieku podścieliska, silnie ekspresjonowanymi genami mezenchymalnymi, aktywacją transformującego czynnika wzrostu beta, gorszym ogólnym i wolnym od nawrotów przeżyciem w porównaniu z pacjentami z innych grup ( 7 , 9 ). Klasyfikacje te dostarczyły informacji na temat właściwego doboru leczenia i rokowania pacjentów, co jest bardzo ważne dla trwających i przyszłych badań klinicznych.
Główne opcje terapeutyczne dostępne obecnie dla pacjentów z CRC to chirurgia, chemioterapia, immunoterapia, radioterapia. 5-letni wskaźnik przeżycia pacjentów z wczesnymi stadiami CRC wynosi prawie 90%. Ze względu na subtelne objawy, u ponad połowy pacjentów diagnozuje się chorobę, gdy rozwinęły się już zaawansowane nowotwory złośliwe. 5-letni wskaźnik przeżycia wynosi tylko 10% lub mniej, gdy pacjenci mają przerzuty ( 10 ).
Wśród nowych potencjalnych podejść terapeutycznych wykazano, że leczenie kannabinoidami i ekstraktami z Cannabis sativa jest skuteczne w hamowaniu wzrostu raka in vitro i in vivo ( 11 ). Roślina C. sativa zawiera fitokannabinoidy, terpenoidy, flawonoidy, kwasy tłuszczowe i inne cząsteczki. Kannabinoidy działają poprzez układ endokannabinoidowy, który składa się z receptorów, takich jak kannabinoid 1 (CB1), kannabinoid 2 (CB2), kanały potencjału receptora przejściowego podtypu waniloidowego 1 i 2 (TRPV1, TRPV2), receptory sprzężone z białkiem G 18, 55, 119 (GPR18, GPR55, GPR119), endokannabinoidy, takie jak 2-arachidonoiloglicerol i anandamid (2-AG, AEA) oraz enzymy odpowiedzialne za ich metabolizm. Głównymi enzymami biosyntetycznymi są NAPE-fosfolipaza D (NAPE-PLD) i lipaza diacyloglicerolowa (DAGL); głównymi enzymami degradacji są hydrolaza amidu kwasu tłuszczowego (FAAH) i lipaza monoacyloglicerolowa (MAGL). Główną funkcją układu endokannabinoidowego jest utrzymanie homeostazy ( 12 ). Receptor CB1 jest głównie ekspresowany w ośrodkowym układzie nerwowym, a receptor CB2, będący najbardziej rozpowszechnionym w układzie odpornościowym, występuje głównie w narządach obwodowych. Oba receptory są receptorami powierzchniowymi komórek sprzężonymi z białkiem G, które są sprzężone ze szlakami cyklazy adenylowej i kinazy białkowej cAMP A oraz szlakami MAPK i PI3K ( 13 ).
Znaczenie układu odpornościowego w raku jelita grubego
W przeszłości guzy były definiowane jako zbiór jednorodnych komórek nowotworowych. Agresywność nowotworu opisywano na podstawie jego cech kliniczno-patologicznych. Ostatnie postępy w immunologii i biologii molekularnej pozwoliły nam lepiej poznać podstawowe mechanizmy potencjału przerzutowego guzów. Wiele badań w tej dziedzinie poszerzyło wiedzę i podkreśliło znaczenie układu odpornościowego w regulacji wzrostu nowotworu. Głównymi graczami tego procesu są wrodzone komórki odpornościowe, takie jak neutrofile, makrofagi, komórki tuczne, eozynofile, komórki supresorowe pochodzące z mieloidu (MDSC) oraz adaptacyjne komórki odpornościowe, takie jak limfocyty T i B ( 14 , 15 ).
W ciągu ostatniej dekady wiedza na temat mikrośrodowiska guza (TME) stała się kluczem do zrozumienia złożonej, wieloetapowej tumorigenezy i opracowania nowych schematów leczenia i leków ( 16 ). Mikrośrodowisko raka obejmuje komórki rezydentne i nierezydentne, które są połączone ze sobą różnymi mediatorami, a każda z nich ma określoną funkcję. Komunikacja między tymi komórkami a komórkami nowotworowymi w ich otoczeniu zasadniczo reguluje los progresji guza. Komórki odpornościowe mogą hamować lub sprzyjać wzrostowi guza ( Tabela 1 ). Nowe badania przedkliniczne wykazały, że atypowe komórki nieprezentujące antygenu są najpierw atakowane przez wrodzony układ odpornościowy; następnie odpowiedź zapalna promuje tworzenie nowych naczyń krwionośnych i proliferację komórek nowotworowych. Niestety, guzy mogą włączać mechanizmy immunosupresyjne i unikać immunonadzoru gospodarza. Adaptacyjna odpowiedź immunologiczna wymaga identyfikacji antygenów nieswoistych poprzez komunikację między białkami a głównym kompleksem zgodności tkankowej komórek prezentujących antygen i receptorami komórek CD8+ i CD4+ T poprzez prezentację antygenu. Guzy mogą utracić swoją antygenowość z powodu nabytych wad w prezentacji antygenu lub mogą zostać zidentyfikowane jako własne ( 25 – 27 ).
Tabela 1.
Działanie pronowotworowe i przeciwnowotworowe komórek układu odpornościowego.
Komórki odpornościowe
Rola w nowotworach (przeciwnowotworowa i pronowotworowa)
Odniesienia
Komórki dendrytyczne (DC)
Uwolnij cytotoksyczne cytokiny Prezentacja antygenu limfocytom T
Wspomaganie wzrostu nowotworu poprzez stymulację neoangiogenezy, przebudowę tkanek i modulację odpowiedzi immunologicznej gospodarza
Istnieją trzy fazy immunoedycji guza: eliminacja, równowaga i ucieczka ( Rysunek 1 ). W pierwszej fazie komórki układu odpornościowego eliminują komórki nowotworowe, które ekspresują białka powierzchniowe. W fazie równowagi niektóre komórki utrzymują się dzięki swojemu potencjałowi do kamuflowania cząsteczek powierzchniowych lub poprzez tłumienie makrofagów i komórek T poprzez ekspresję substancji takich jak PD-1/2 na komórkach prezentujących antygen. W ostatniej fazie niektóre komórki mogą uniknąć zabicia, co następnie prowadzi do ucieczki i proliferacji opornych klonów. Ponadto degradacja macierzy zewnątrzkomórkowej przez metaloproteinazy macierzy i nowe naczynia krwionośne utworzone w wyniku nieprawidłowej angiogenezy sprzyjają tworzeniu się przerzutów ( 15 ).
Rysunek 1.
Fazy immunoedycji w raku jelita grubego. Eliminacja obejmuje usunięcie komórek nowotworowych, równowaga opisuje przeżycie frakcji przekształconych komórek, a ucieczka opisuje ucieczkę i proliferację tych komórek.
Jeśli chodzi o ekspresję receptorów kannabinoidowych w komórkach układu odpornościowego, wykazano, że receptory te są wyrażane zarówno w odporności adaptacyjnej, jak i wrodzonej. Na przykład receptory CB1, CB2 i GPR55 są wyrażane na komórkach NK, CB1, CB2 – na komórkach tucznych, limfocytach T – na komórkach B. Dlatego można postawić hipotezę, że fitokannabinoidy mogą wpływać na funkcjonowanie układu odpornościowego, regulować stan zapalny i posiadać działanie przeciwnowotworowe itp. ( 28 ).
Rola stanu zapalnego w karcynogenezie jelita grubego
Zapalenie odgrywa kluczową rolę w karcynogenezie jelita grubego i jest obecnie uważane za jedną z pojawiających się cech charakterystycznych raka ( 29 ). Lepsze zrozumienie CRC i zapalenia może doprowadzić do opracowania nowych biomarkerów nowotworowych i bardziej spersonalizowanych i skutecznych terapii. Wiadomo, że pacjenci cierpiący na przewlekłe schorzenia, takie jak choroba zapalna jelit, mają znacznie wyższe ryzyko rozwoju CRC ( 30 ). Zapalenie jest uważane za ważną siłę napędową nowotworów CRC związanych z zapaleniem jelita grubego, podczas gdy jego rola w przypadku nowotworów sporadycznych i dziedzicznych jest mniej jasna. Dowody wskazują, że niesteroidowe leki przeciwzapalne mogą zapobiegać rozwojowi CRC lub go opóźniać ( 31 ). Metaanaliza badań z randomizacją wykazała, że podczas obserwacji po 20 latach stosowania aspiryny przez 5 lat śmiertelność i częstość występowania CRC uległyby zmniejszeniu o 30–40% ( 32 ).
Na podstawie klasyfikacji CMS CRC, CMS1 i CMS4 są uważane za zapalne, przy czym pierwszy ma złe rokowanie po nawrocie, a drugi — najgorszy wskaźnik przeżycia. Ogólnie rzecz biorąc, stan zapalny odgrywa podwójną rolę w nowotworze. Celowanie w komórki złośliwe przez cytotoksyczne limfocyty T lub zmniejszanie niespecyficznego stanu zapalnego przez T-regs może prowadzić do odpowiedzi przeciwnowotworowej. Ten typ odpowiedzi nazywa się ochronną i jest związany z polaryzacją Th1 i niższym nawrotem CRC. Podtyp Th1 wytwarza IFN-γ i wzmacnia toksyczność komórkową, podczas gdy podtyp Th2 uwalnia IL-4 i wzmacnia humoralną odpowiedź komórek B. Najczęstszymi cytokinami prozapalnymi są TNF-α, IL-6, IL-12, IL-2, a najczęstszymi przeciwzapalnymi są IL-10, IL-4, IL-5, TGF-β i IFN-α ( Tabela 2 ). Komórki odporności wrodzonej i nabytej oraz inne komórki, takie jak fibroblasty, komórki mezenchymalne i perycyty, odgrywają ważną rolę w zapaleniu związanym z rakiem ( 57 ). Komunikacja między tymi komórkami odbywa się za pośrednictwem sieci cytokin produkowanych i wydzielanych przez komórki układu odpornościowego po stymulacji. Rola szlaku sygnałowego IL-10 pozostaje kontrowersyjna w przypadku CRC. Wyższy poziom IL-10 wiąże się z gorszym przeżyciem pacjentów, podczas gdy badania na zwierzętach pokazują, że pełni on rolę ochronną poprzez tłumienie stanu zapalnego ( 46 , 47 ). IL-6 jest aktywatorem szlaku sygnałowego STAT-3 i często występuje u pacjentów z CRC; i jest również powiązany z gorszym przeżyciem i zwiększonym ryzykiem nawrotu ( 37 , 57 , 58 ). Fibroblasty podścieliska, uzyskane z raka jelita grubego, wyprodukowały znaczne ilości IL-6. Ostatni z nich indukował angiogenezę guza poprzez zwiększenie produkcji VEGF ( 38 ). IL-6 ułatwia kolonizację przerzutową komórek raka jelita grubego. U myszy IL6-/- przerzuty komórek CT26 do wątroby zostały zmniejszone, a funkcja komórek CD8+ T uległa poprawie in vivo . Co więcej, myszy z niedoborem IL-6 skutecznie odpowiedziały na wstrzyknięcie anty-PD-L1 poprzez zahamowanie kolonizacji przerzutowej, podczas gdy u myszy IL6+/+ nie zaobserwowano tego efektu ( 39 ). IFN-γ jest wytwarzany przez komórki CD4+, CD8+ i NK i indukuje apoptozę komórek. Utrata jednej kopii tego interferonu u myszy Apcmin/+ wykazała znacznie szybszy postęp w kierunku gruczolakoraka jelita grubego. Komórki CRC mogą minimalizować przeciwnowotworowe efekty sygnalizacji interferonu przez łańcuch receptora interferonu typu I, co prowadzi do słabej odpowiedzi na inhibitory punktów kontrolnych anty-PD1 ( 44 ). Ekspresja TNF-α jest znacznie wyższa w raku jelita grubego niż w sąsiadującej prawidłowej tkance jelita grubego. Zwiększona ekspresja tej cytokiny silnie koreluje z bardziej zaawansowanymi nowotworami ( 59). Po stymulacji TNF-α zauważono wzrost onkogenu Metastasis-Associated in Colon Cancer 1 (MACC1) zarówno na poziomie mRNA, jak i białka. MACC1 indukuje proliferację, przeżycie i przerzuty komórek nowotworowych. Poziomy ekspresji tego onkogenu zostały zmniejszone przez obniżenie p65 NF-kB. Ponadto indukcję MACC1 utrudniało monoklonalne przeciwciało anty-TNF-α, adalimumab ( 34 ). TNF-α zwiększał poziomy cytokin prozapalnych, takich jak IL-6 i IL-8 in vitro na komórkach raka jelita grubego HT-29 ( 35 ). Inne badanie wykazało, że wpływ szczepionki peptydowej, AH1, na myszy z guzem jelita grubego CT26 spowodował skromne zahamowanie wzrostu guza, ale połączenie z F8-TNF drastycznie zwiększyło aktywność przeciwnowotworową. F8-TNF to białko fuzyjne przeciwciał, które dostarcza TNF do macierzy zewnątrzkomórkowej guza. Synergia między szczepionką peptydową a białkiem fuzyjnym TNF została wyjaśniona tym, że F8-TNF powoduje szybką martwicę krwotoczną guza i w rezultacie pozostawia niewielką ilość resztkowych komórek nowotworowych. Ponadto zauważono znaczący wzrost specyficznych dla AH1 komórek CD8+ T w guzach i drenujących węzłach chłonnych ( 60 ). IL-12 hamowała ruchliwość i inwazję ludzkich komórek raka jelita grubego (HRT18, HT29 i HT115), co sugeruje jej ważną rolę w przerzutach ( 41 ). IL-4 jest aktywnie uwalniana przez komórki macierzyste raka jelita grubego i nadaje guzom fenotyp odporny na śmierć. Neutralizacja IL-4 za pomocą przeciwciała znacząco uwrażliwia komórki nowotworowe na chemioterapię ( 49 ). Wczesna transgeneza IL-5 w mysim modelu CRC z zapaleniem jelita grubego zwiększyła ciężkość zapalenia jelita grubego, wywołała szybkość tworzenia polipów i w rezultacie większe obciążenie guzem ( 51 ). U pacjenta zgłoszono przypadek skrajnej eozynofilii spowodowanej przez rozsianego raka jelita grubego wytwarzającego IL-5 ( 61 ). TGF-β promuje przeżycie, inwazję i przerzuty komórek CRC ( 53 ). TGB-β w mikrośrodowisku guza zwiększa wykluczenie komórek T i hamuje wykopywanie fenotypu Th-1. Myszy z przerzutowym rakiem jelita grubego i zablokowaną ścieżką sygnałową TGF-β mają guzy wrażliwe na terapię anty-PD-1 anty-PD-L1. Natomiast myszy z odblokowaną sygnalizacją TGF-β wykazały ograniczoną odpowiedź na inhibitory punktów kontrolnych układu odpornościowego ( 54 ). Systemowe podanie IFN-α myszom z rakiem jelita grubego znacząco zahamowało wzrost guza i jego unaczynienie; indukowanej apoptozy komórek nowotworowych i komórek śródbłonka wątroby związanych z przerzutami ( 56 ).
Tabela 2.
Główne efekty cytokin pro- i przeciwzapalnych.
Cytokiny
Miejsce produkcji
Ruchomości
Znaczenie dla karcynogenezy jelita grubego
Odniesienia
Cytokiny prozapalne
TNF-alfa
Makrofagi, limfocyty T, komórki NK, komórki tuczne, eozynofile
Stymulacja stanów zapalnych, odporność na infekcje i nowotwory
TNF-α reguluje indukcję MACC1 poprzez podjednostkę NF-κB p65. Zwiększa poziom IL-6 i IL-8.
Optymalny sposób leczenia można wybrać, łącząc i analizując informacje na temat czynników związanych z nowotworem (lokalizacja nowotworu, obecność przerzutów, obecność biomarkerów itp.) oraz czynników związanych z pacjentem (rokowanie, choroby współistniejące itp.).
Pacjenci z rakiem jelita grubego z chorobą przerzutową otrzymują kombinację chemioterapii i immunoterapii. Chemioterapia pierwszego rzutu obejmuje fluoropirymidyny, takie jak kapecytabina i 5-fluorouracyl (5-FU) w monoterapii lub z leukoworyną (LV), oksaliplatyną (5-FU/LV/oksaliplatyna – FOLFOX), irynotekanem (5-FU/LV/irynotekan – FOLFIRI), kapecytabiną/LV/oksaliplatyną – CAPOX. Chemioterapia drugiego rzutu – FOLFOX lub CAPOX dla pacjentów opornych na irynotekan. Pacjentom opornym na kombinacje oksaliplatyny przepisuje się FOLFIRI lub irynotekan w monoterapii. Zwykle leczenie trwa do 6 miesięcy, ale czas trwania w znacznym stopniu zależy od indywidualnych przypadków ( 62 ).
Najczęstsze działania niepożądane chemioterapii w przypadku CRC to leukopenia, polineuropatia, biegunka, trombocytopenia, nadmierne wymioty, dysfunkcje wątrobowo-nerkowe i pogorszenie ogólnego stanu. Nasilenie działań niepożądanych jest zwykle większe u pacjentów w podeszłym wieku i u pacjentów z wcześniej istniejącymi chorobami współistniejącymi ( 63 ). Ze względu na obawy dotyczące toksyczności chemioterapia może nie być odpowiednia dla wielu pacjentów. Onkolodzy mogą nie zalecać tego typu leczenia ze względu na niektóre zaawansowane stadia chorób przewlekłych (niewydolność wątroby, nerek i serca) i słabą sprawność fizyczną ( 64 ).
Immunoterapia
Immunoterapia jest jedną z najbardziej obiecujących metod terapeutycznych dla pacjentów z rakiem jelita grubego ( 65 ). Terapia celowana zrewolucjonizowała leczenie raka. Immunoterapia jest rodzajem podejścia leczniczego, które pomaga układowi odpornościowemu w eliminacji guzów. Można ją podzielić na dwie główne grupy: aktywną (szczepionki) i pasywną (przeciwciała monoklonalne, adoptywna terapia komórkowa) ( Tabela 3 ). Ponadto niektóre terapie biologiczne mogą być szczególnie ukierunkowane na określone antygeny nowotworowe, podczas gdy inne działają niespecyficznie, wzmacniając naturalne odpowiedzi immunologiczne ( 75 ).
Tabela 3.
Zalety i wady środków immunoterapeutycznych.
Immunoterapia
Zalety i wady
Status zatwierdzenia w CRC
Odniesienia
Szczepionki na cały guz
Składa się ze wszystkich znanych i nieznanych antygenów nowotworowych, łatwa produkcja
Istnieją pewne rodzaje szczepionek przeciwnowotworowych, które badano w leczeniu CRC, takie jak szczepionki na cały guz, peptydy, wektory wirusowe i komórki dendrytyczne (DC). Celem tych środków, podobnie jak każdej innej strategii immunizacji, jest wywołanie odpowiedzi immunologicznej przeciwnowotworowej, która wyeliminuje raka i zapewni organizmowi ciągły nadzór w celu ochrony przed jego nawrotem.
Szczepionki na cały guz
Niektóre zalety pracy ze szczepionkami na cały guz to: są łatwe w produkcji i składają się ze wszystkich znanych i nieznanych antygenów nowotworowych. Natomiast najistotniejszą wadą tych szczepionek jest bardzo niska immunogenność, która może być skierowana do normalnych komórek, a w rezultacie niska skuteczność. Podjęto kilka podejść w celu zwiększenia immunogenności szczepionek na cały guz. Przeprowadzono badanie z zakażeniem wirusem choroby Newcastle, które wykazało 98% 2-letni wskaźnik przeżycia u pacjentów z wyciętym rakiem jelita grubego w porównaniu z 67% u pacjentów, którzy otrzymali szczepionkę na cały guz w połączeniu ze szczepionką Bacillus Calmette-Guérin (BCG). Wyniki sugerują, że immunogenność tych związków została poprawiona ( 27 ).
Szczepionki peptydowe
Szczepionki peptydowe są bardziej specyficzne dla antygenu związanego z guzem, ale ich skuteczność jest nadal znacznie niska ze względu na niewielką ilość odpowiedzi komórek T. Badanie fazy I/II przeprowadzone u pacjentów z rakiem jelita grubego wykazało, że połączenie szczepionki p53 z interferonem alfa zwiększyło ilość interferonu gamma ( 76 ). Następny typ szczepionki to szczepionki wektorowe wirusowe. Są specyficzne dla antygenu związanego z guzem i naturalnie immunogenne. Wadą ich stosowania jest ich zdolność do wywoływania burzy cytokinowej. Najczęściej stosowanymi wirusami w raku jelita grubego są adenowirusy, pokswirusy i alfawirusy. Większość z tych szczepionek jest ukierunkowana na antygen karcinoembrionalny (CEA), białko wyrażane przez raki jelita grubego. Dane przedkliniczne pokazują, że rekombinowany wirus ospy wyrażający CEA (rV-CEA) może wzmacniać adaptacyjne i wrodzone odpowiedzi immunologiczne u myszy. Ponadto hamował proliferację gruczolakoraka jelita grubego w modelach zwierzęcych. Jednakże badania kliniczne przeprowadzone na pacjentach w zaawansowanym stadium raka jelita grubego wykazały brak odpowiedzi ( 77 ). Szczepionki z komórek dendrytycznych charakteryzują się specyficznością antygenu związanego z guzem i generowaniem własnej odpowiedzi immunologicznej organizmu. Negatywnymi aspektami są wysokie koszty i bardzo czasochłonny proces przygotowania. Po całkowitym wycięciu przerzutów CRC do wątroby, badanie kliniczne szczepionki fazy II wykazało mniej i opóźnione nawroty w grupie szczepionej w porównaniu z grupą obserwacyjną ( 78 ). Wyniki szczepionek DC są bardzo zachęcające, a wkrótce ich skuteczność może zostać znacznie poprawiona.
Terapia Adopcyjnego Transferu Komórek
Adopcyjna terapia transferu komórek to kolejny rodzaj immunoterapii. Głównymi zaletami tego typu leczenia są eliminacja konieczności wytworzenia odpowiedzi immunologicznej i wysoka swoistość guza. Z kolei pewnymi wadami są wysokie koszty, długi czas przygotowania i toksyczność zależna od celu. W tej terapii autologiczne komórki T są pobierane z guza, węzłów chłonnych lub krwi obwodowej pacjenta i modyfikowane ex vivo poprzez ich ekspansję i dodanie niektórych cząsteczek współstymulujących i cytokin. Następnie przeprowadza się bierny transfer tych komórek T do gospodarza w celu bezpośredniego zniszczenia guza. Najnowszym odkryciem tego typu biernej immunoterapii jest opracowanie zmodyfikowanych komórek T, które wyrażają chimeryczne receptory antygenowe specyficznie dla antygenu karcinoembrionalnego ( 79 ). Badanie fazy I przeprowadzone u pacjentów z rakiem jelita grubego opornym na standardowy schemat protokołu leczenia przy użyciu autologicznych komórek T zmodyfikowanych w celu wyrażenia receptora komórek T CEA myszy wykazało znaczący spadek stężenia CEA w surowicy u wszystkich trzech pacjentów; u jednego z nich wykazano odpowiedź kliniczną w postaci regresji przerzutów w wątrobie i płucach. Jednocześnie u tych pacjentów wystąpiło przejściowe zapalenie jelit ( 80 ). Niedawno opublikowane wyniki badania przypadku u pacjentów z zaawansowanym rakiem jelita grubego wykazały znaczącą odpowiedź kliniczną na połączenie kapecytabiny i adopcyjnego transferu komórek (komórek αβT i komórek NK) przepisanych po laparoskopowej resekcji raka jelita grubego i niektórych przerzutów do wątroby. Dwa tygodnie po laparoskopii zaobserwowano drastyczny wzrost poziomu CEA. Adopcyjny transfer komórek pozwolił na obniżenie poziomu CEA w surowicy, ostatecznie doprowadzając go do normy. Zaobserwowano zauważalną redukcję wielkości nieoperacyjnych przerzutów do wątroby. Podczas badania kontrolnego po 19 miesiącach nie odnotowano progresji ani nawrotu, a poziomy CEA utrzymywały się w granicach normy ( 81 ).
Immunoterapia oparta na przeciwciałach
Wysoce specyficzne przeciwciała monoklonalne są bardzo skuteczne w leczeniu raka od dziesięcioleci. Białka przeciwko receptorowi naskórkowego czynnika wzrostu (EGFR) i czynnikowi wzrostu śródbłonka naczyniowego (VEGF) w połączeniu z chemioterapią wykazały lepsze wyniki leczenia złośliwego CRC (mCRC). Środki anty-EGFR, takie jak Cetuximab lub Panitumumab, w monoterapii lub w połączeniu z lekami cytotoksycznymi są przepisywane tylko wtedy, gdy nie ma mutacji KRAS ( 62 ). Bevacizumab, humanizowane przeciwciało monoklonalne przeciwko VEGF, hamuje wzrost guza i angiogenezę, a także moduluje układ odpornościowy gospodarza poprzez zwiększenie populacji komórek B i T ( 82 ).
Dramatyczną skuteczność immunoterapii opartej na przeciwciałach udowodniono przy użyciu innego typu przeciwciał monoklonalnych (mAbs) znanych jako inhibitory punktów kontrolnych (ICI). Wykazano, że obecnie stosowane ICI zapewniają znaczące odpowiedzi kliniczne u pacjentów z mCRC, szczególnie z typem niedoboru naprawy niedopasowania/wysokiej niestabilności mikrosatelitarnej (dMMR-MSI-H). Celują one w hamujące receptory immunologiczne: programowaną śmierć komórki 1 (PD-1) i jej ligand PD-L1, cytotoksyczny antygen związany z limfocytami T 4 (CTLA-4). Ten ostatni jest wyrażany w naiwnych limfocytach T, efektorowych limfocytach T i regulatorowych limfocytach T (T-regs). Stymuluje on dezaktywację T-regs poprzez wiązanie się z komórkami prezentującymi antygen. Receptor PD-1 jest obecny w limfocytach CD4, CD8, komórkach NK, MDSC, T-regs i limfocytach B. Razem ze swoim ligandem, ten receptor powoduje wyczerpanie komórek T poprzez minimalizowanie naciekających guz limfocytów i proliferacji komórek T. W konsekwencji, guzy nabywają immunooporności. Środki anty-CTLA-4 i anty-PD-1 aktywują komórki T i powodują silniejszą odpowiedź przeciwnowotworową ( 83 ). Guzy CRC dMMR-MSI-H mają 20 razy większy ładunek mutacji niż guzy z niestabilnością mikrosatelitarną o niskiej sprawności naprawy niezgodności (pMMR-MSI-L). Ponadto są bardziej naciekane przez TIL, makrofagi i mają podwyższone poziomy cytokin stymulujących układ odpornościowy w porównaniu z pMMR-MSI-L. Ten ostatni ma mniej skuteczną odpowiedź na ICI i gorsze rokowanie ( 84 ).
W sierpniu 2020 r. istniały trzy zatwierdzone przez FDA ICI, które były stosowane u pacjentów z dMMR-MSI-H mCRC. Pierwszym z nich był Nivolumab, lek przeciwko PD-1 zatwierdzony przez FDA w lipcu 2017 r. po pomyślnych wynikach badania fazy II CheckMate 142 dotyczącego leczenia drugiej linii pacjentów z dMMR-MSI-H mCRC, u których wystąpił postęp choroby podczas leczenia oksaliplatyną, fluoropirymidyną i irynotekanem. W badaniu tym zgłoszono, że po 12 miesiącach obserwacji obiektywny wskaźnik odpowiedzi wystąpił u 31% pacjentów i 69% osób z grupy kontrolnej. 12-miesięczne przeżycie bez progresji (PFS) wyniosło 50%, a całkowite przeżycie (OS) 73%. Najczęstszymi działaniami niepożądanymi związanymi z terapią były świąd, wysypka, biegunka i zmęczenie. Mutacje BRAF, KRAS i ekspresja PD-L1 nie miały wpływu na odpowiedź na przepisaną terapię celowaną ( 85 ).
Drugim ICI zatwierdzonym przez FDA w maju 2017 r. był Pembrolizumab, substancja przeciwko PD-1, której skuteczność została udowodniona w pierwszej fazie badania Keynote 016. Początkowo wykazano, że pacjenci z mCRC dMMR-MSI-H doświadczyli 40% wskaźnika odpowiedzi (RR), podczas gdy pacjenci z pMMR-MSI-L – 0% RR. Później udokumentowano, że 2-letni PFS wynosił 53% w pierwszej grupie. Ciężkie działania niepożądane wystąpiły tylko u 14% pacjentów, w tym trombocytopenia, leukopenia i zapalenie trzustki. Ta lecznicza opcja monoterapii została przepisana pacjentom z dMMR-MSI-H mCRC, których stan pogorszył się w trakcie lub po terapii oksaliplatyną, fluoropirymidyną i irynotekanem.
Następnym podejściem immunoterapeutycznym zatwierdzonym przez FDA w przypadku opornego CRC, który rozwinął się podczas terapii oksaliplatyną, fluoropirymidyną i irynotekanem, było połączenie niwolumabu z ipilimumabem (środek anty-CTLA-4). Zatwierdzenie zostało przyznane w lipcu 2018 r., po raporcie wyników badania fazy II CheckMate 142. Podczas obserwacji po 13,4 miesiącach obiektywny wskaźnik odpowiedzi wyniósł 54,7%, przy częściowej odpowiedzi −51,3%, całkowitej odpowiedzi −3,4%, a wskaźnik kontroli choroby przez 3 miesiące lub dłużej −80%. PFS po 12 miesiącach wyniósł 71%, OS −85%. Trzynaście procent pacjentów było zmuszonych przerwać leczenie z powodu działań niepożądanych związanych z lekiem. Ta kombinacja wykazała lepszą skuteczność niż monoterapia anty-PD-1. Jednakże działania niepożądane stopnia 3–4 były bardziej widoczne w przypadku terapii skojarzonej w porównaniu z leczeniem jednym lekiem i wynosiły odpowiednio 32–20% ( 86 ).
W przypadku pacjentów z pMMR-MSI-L należy przeprowadzić więcej badań nad schematami immunoterapeutycznymi. Istnieje potrzeba znalezienia leków, które będą ukierunkowane na odpowiedź immunologiczną, a także będą promować naciek komórek T. Ze względu na niskie obciążenie mutacjami i neoantygenami trudno jest osiągnąć te cele. Obecne schematy obejmują radioterapię, chemioterapię i substancje antyangiogenne w celu zwiększenia aktywacji immunologicznej, zabijania komórek nowotworowych i podwyższenia antygenów nowotworowych. Później leczenie można łączyć z ICI i innymi lekami biologicznymi. Obecnie trwają pewne badania kliniczne, które oceniają skutki chemioterapii z zastosowaniem terapii anty-PD-1, anty-PD-L1 i zewnętrznej radioterapii wiązką lub ablacji częstotliwości radiowej ( 87 ). Trwające badanie NCT01633970 fazy Ib oceniało skuteczność atezolizumabu (anty-PD-L1) i bevacizumabu plus FOLFOX; wykazało ono OS na poziomie 7% i stabilizację choroby u 64% pacjentów. Innym dobrze przebadanym podejściem jest połączenie inhibitorów kinazy białkowej aktywowanej mitogenem (MEK), takich jak Cobimetinib i Atezolizumab. Inhibitory MEK mogą dodatkowo uwrażliwić MSS mCRC na terapię celowaną. Badanie kliniczne fazy Ib ( NCT01988896 ) oceniło to połączenie u pacjentów z opornym na leczenie mutantem KRAS CRC i pMMR-MSI-L CRC i wykazało RR wynoszący 17%, gdzie pięciu pacjentów z 23 miało stabilną chorobę, a u czterech pacjentów rozwinęła się PR. Nie odnotowano żadnych zaawansowanych działań niepożądanych związanych z terapią. Później włączono 84 pacjentów, a wyniki zaktualizowano. RR wyniosło 8%, wskaźnik kontroli choroby -−31%. 6-miesięczne PFS i 12-miesięczne OS wyniosły odpowiednio 27 i 51%. To podejście jest bardzo obiecujące, ponieważ pokazuje, że inhibitory MEK mogą zwiększyć odpowiedź na immunoterapię u pacjentów z MSS mCRC. Podczas trwającego badania NCT03406871 , w którym połączono niwolumab i regofarenib (inhibitor wielokinazowy), przedstawiono obiecujące wyniki ; u 18 z 19 pacjentów wystąpiła obiektywna odpowiedź guza (siedem z nich to MSS CRC, 11 — rak żołądka MSS i 1 — MSI-H CRC). Nadal konieczne są bardziej spersonalizowane podejścia do leczenia pMMR-MSI-L ( 84 ).
Znaczenie ECS dla CRC
ECS aktywnie reguluje homeostazę jelit. Wszystkie składniki ECS są silnie wyrażone w tkance jelitowej, co oznacza, że ten system bezpośrednio wpływa na prawidłowe funkcjonowanie układu żołądkowo-jelitowego. Receptory CB1 i CB2 są wyrażone w zdrowym nabłonku okrężnicy, podśluzówkowym splocie mięśniowo-jelitowym i mięśniach gładkich, komórkach plazmatycznych w blaszce właściwej; receptor CB2 jest również obecny na makrofagach jelitowych ( 88 , 89 ). Receptor TRPV1 jest wyrażany na włóknach nerwowych okrężnicy ( 90 ). Receptor GPR55 jest obecny w błonie śluzowej i warstwie mięśniowej okrężnicy ( 91 ). Endokannabinoidy, 2-AG i AEA są również obecne w zdrowej tkance okrężnicy ( 92 ). Główne enzymy degradujące endokannabinoidy, enzymy FAAH i MAGL są rozmieszczone na gruczołach nabłonka okrężnicy, blaszce właściwej i splocie mięśniowo-jelitowym. Enzymy biosyntezy NAPE-PLD i DAGL występują na mięśniach gładkich jelita grubego, blaszce właściwej i gruczołach nabłonkowych; DAGL występuje również na splocie mięśniówkowym jelita ( 89 ).
Aby zrozumieć rolę ECS w jelitach, ważne jest rozróżnienie skutków zwiększonego i zmniejszonego tonu kannabinoidowego w układzie żołądkowo-jelitowym. Ogólnie rzecz biorąc, antagoniści receptora CB1 zmniejszają ton kannabinoidowy w jelitach i prowadzą do wymiotów, biegunki, zwiększonego opróżniania żołądka i pasażu żołądkowo-jelitowego. Natomiast agoniści receptora CB1 i CB2, a także inhibitory MAGL i blokery FAAH prowadzą do zwiększenia tonu kannabinoidowego jelit poprzez zmniejszenie wymiotów, wydzielania kwasu żołądkowego i opróżniania żołądka, a także zmniejszenie nadruchliwości, biegunki i bólu trzewnego ( 93 ). Wyciszenie receptora CB1 przez selektywnego antagonistę receptora CB1 AM251 u myszy ApcMin/+ doprowadziło do zwiększenia liczby polipów jelitowych, podczas gdy aktywacja receptora CB1 spowodowała śmierć komórek nowotworowych. Natomiast wyciszenie receptora CB2 nie wykazało żadnego wpływu na wzrost polipów ( 94 ).
Składniki ECS są znacząco rozregulowane w CRC. Endokannabinoidy (2-AG i AEA) były 3-krotnie wyższe w gruczolakach i 2-krotnie wyższe w CRC w porównaniu do prawidłowej błony śluzowej jelita grubego ( 92 ). Ekspresja receptora CB1 jest zmniejszona w CRC ( 95 ). Ekspresja receptora CB2 jest zwiększona w CRC i jest uważana za zły czynnik prognostyczny w tym typie raka ( 96 ). Poziomy FAAH i MAGL były również zwiększone u pacjentów z CRC ( 97 ). ECS jest bardzo ważnym czynnikiem patogenezy CRC, co sugeruje potencjalny wpływ kannabinoidów na tę chorobę.
Roślina lecznicza , która ostatnio zyskała wiele uwagi w dziedzinie leczenia raka, to Cannabis sativa . Wiele eksperymentów in vitro i in vivo wykazało, że kannabinoidy i ekstrakty z konopi hamują proliferację, stymulują apoptozę i autofagię, tłumią angiogenezę i przerzuty ( 98–100 ). Głównymi aktywnymi kannabinoidami odpowiedzialnymi za te efekty są kannabigerol (CBG), kannabidiol (CBD) i tetrahydrokannabinol (THC). Wykazano, że CBG aktywował apoptozę, stymulował produkcję ROS, zwiększał mRNA CHOP i hamował wzrost komórek w komórkach CRC (Caco-2, HCT-116) ( 101 ). Stwierdzono, że hamujący wpływ CBG na żywotność komórek raka jelita grubego zależał od czasu. W wyciszonych komórkach TRPM8 hamujący wpływ CBG na wzrost komórek był wyraźnie tłumiony w porównaniu z komórkami niewyciszonymi. Indukcję apoptozy wykazano poprzez wzrost aktywności kaspaz 3 i 7, obecność fragmentów DNA, wzrost ekspresji CHOP. W tym samym artykule wykazano, że CBG (3 lub 10 mg/kg) hamowało wzrost guzów ksenoprzeszczepowych (HCT-116) w modelu myszy o 45,3% i chemicznie indukowało karcynogenezę jelita grubego w modelach za pomocą azoksymetanu (AOM), w którym CBG w stężeniu 5 mg/kg całkowicie hamowało powstawanie nieprawidłowych ognisk krypt (ACF), zmniejszało liczbę guzów o połowę i nie wpływało na powstawanie polipów ( 101 ).
Wykazano również, że CBD ma działanie antyproliferacyjne w modelach raka jelita grubego. W niektórych badaniach in vitro CBD chroniło DNA przed stresem oksydacyjnym, podnosiło poziom endokannabinoidów i hamowało proliferację komórek raka jelita grubego za pośrednictwem receptorów CB1, TRPV1, PPAR-γ ( 102 ). Selektywni antagoniści rimonabant i AM251 (antagonista CB1R), kapsazepina (antagonista TRPV1R), GW 9662 (antagonista receptora PPAR-γ) hamowali działanie antyproliferacyjne CBD. Chemoprewencja CBD została potwierdzona przy użyciu modeli in vivo raka jelita grubego wywołanego AOM. CBD (1 mg/kg) zmniejszyło ACF o 67%, liczbę guzów o 66% i polipów o 57%. Gdy stężenie wzrosło do 5 mg/kg, zapobiegało jedynie tworzeniu się polipów. Efekt ten był spowodowany aktywacją kaspazy-3 i zmniejszeniem fosforylowanej formy białka Akt ( 102 ). W innym badaniu wykazano proapoptotyczny efekt CBD w komórkach CRC (HCT-116, DLD-1) i zasugerowano, że jest on wynikiem aktywacji Noxa, zwiększenia produkcji ROS i indukcji stresu siateczki śródplazmatycznej. Gdy poziomy Noxa zostały stłumione przez siRNA, ekspresja markerów apoptozy uległa znacznemu zmniejszeniu. Podobnie, po zablokowaniu produkcji ROS, poziom Noxa został zmniejszony. CBD indukował apoptozę w sposób zależny od Noxa-ROS ( 103 ). Ponadto, podczas stosowania raka jelita grubego indukowanego linią komórkową CT26 u myszy, CBD w stężeniach 1 i 5 mg/kg wykazywało działanie przeciwangiogenne i przeciwprzerzutowe poprzez hamowanie VEGF, przy czym ta druga dawka była skuteczniejsza. U zwierząt otrzymujących CBD zaobserwowano istotny wzrost aktywności enzymów antyoksydacyjnych, w tym SOD, GPX, GR, TAC i spadek MDA ( 104 ).
Badano również wpływ pełnych ekstraktów botanicznych, takich jak botaniczna substancja lecznicza o wysokiej zawartości CBD (BDS), na raka jelita grubego. Takie ekstrakty są zazwyczaj przygotowywane z kwiatów konopi, które są bogate w CBD, lub izolat CBD jest dodawany (wzbogacany) do określonego stężenia. Postawiono hipotezę, że inne składniki ekstraktów z roślin konopi mogą działać synergicznie z CBD i mogą być przydatne z terapeutycznego punktu widzenia. Wykazano, że CBD BDS ma znaczące właściwości antyproliferacyjne na komórki rakowe (HCT-116, DLD-1), podczas gdy zdrowe komórki nabłonka jelita grubego nie zostały dotknięte. Nie odnotowano różnicy w sile i skuteczności między CBD BDS a CBD, gdy stosowano te same dawki (0,3–5 μM). Efekty CBD BDS były neutralizowane przez selektywnych antagonistów receptorów CB1 i CB2. CBD BDS miało wyraźniejsze powinowactwo do receptorów CB1 i CB2 niż czysty CBD. Badania in vivo wykazały, że zastosowanie chemicznie indukowanej karcynogenezy przez AOM, ekstraktu z C. sativa o wysokiej zawartości kannabidiolu, zahamowało ACF o 86%, polipy o 79% i tworzenie się guzów o 40%. W modelach ksenoprzeszczepu CBD BDS znacząco zmniejszyło objętość guza, ale nie zaobserwowano różnicy w rozwoju guzów po 1 tygodniu leczenia ( 105 ).
Wykazano, że THC indukuje apoptozę w komórkach raka jelita grubego poprzez aktywację receptorów CB1 i hamowanie PI3K-AKT, kaskady RAS-MAPK i aktywacji BAD. Komórki raka jelita grubego (SW480, HCT-15, HT29, Caco-2, HCT-116 i SW620) wystawione na działanie THC (2,5–12,5 μM) skutkowały dawkozależnym zmniejszeniem przeżywalności komórek. Natomiast mniejsze stężenia od 100 nM do 1 μM nie miały zauważalnego wpływu na proliferację i przeżywalność komórek raka jelita grubego. THC zwiększało poziomy kaspazy-3 i PARP (substratu kaspazy-3). THC powodowało defosforylację i aktywację BAD ( 106 ). Potencjał przeciwnowotworowy kannabinoidów w CRC podsumowano w Tabeli 4 .
Tabela 4.
Potencjał przeciwnowotworowy kanabinoidów w raku jelita grubego.
Fitokannabinoidy
Typ modelu
Działanie przeciwnowotworowe/mechanizm działania
Odniesienia
CBG
in vitro (Caco-2, HCT116) in vivo (samice myszy grasicy, ksenograft-HCT116; model raka jelita grubego wywołanego azoksymetanem)
Proapoptotyczne, antyproliferacyjne; pobudza produkcję ROS, zwiększa mRNA CHOP, zwiększa poziom aktywności kaspazy 3, 7; w guzach ksenoprzeszczepionych – zmniejsza wzrost guza, w guzach AOM – całkowicie hamuje ACF, zmniejsza liczbę guzów
Powolny rozwój i zatwierdzanie nowych leków przeciwnowotworowych na CRC wynika z braku odpowiednich modeli przedklinicznych. Modele 2D in vitro umożliwiają przeprowadzanie badań przesiewowych o wysokiej przepustowości i są proste w obsłudze, ale pozwalają jedynie na badanie interakcji komórka-komórka lub komórka-matryca, a nie całego TME; dlatego nie są fizjologicznie istotne i nie są klinicznie predykcyjne. Z drugiej strony, modele zwierzęce in vivo pozwalają na badanie interakcji całego organizmu z odpowiednim TME i heterogenicznością wewnątrzguzową, ale te modele nie nadają się do badań przesiewowych na dużą skalę, są bardzo czasochłonne i nie są „ludzkie”. Tak więc zarówno modele in vitro , jak i in vivo stanowią cenne narzędzie do badania karcynogenezy jelita grubego ( 107 ). Jednak ze względu na wspomniane różnice korelacja między tymi modelami nie jest zbyt silna ( 108 ). Z drugiej strony badania kliniczne są złotym standardem testowania i zatwierdzania każdego potencjalnego leku.
Ważne jest, aby wspomnieć o jednym badaniu klinicznym, które zbadało największą liczbę pacjentów onkologicznych otrzymujących medyczną marihuanę w latach 2015-2017 w Izraelu. Dwa tysiące dziewięćset siedemdziesięciu pacjentów cierpiących na raka piersi (20,7%), płuc (13,6%), trzustki (8,1%) i jelita grubego (7,9%) otrzymywało medyczną marihuanę jako leczenie paliatywne w celu złagodzenia objawów, takich jak ból, brak apetytu, złe samopoczucie, zaburzenia snu i nudności. W tym badaniu wykorzystano cztery rodzaje marihuany: szczepy sativa o wysokiej zawartości THC, bez CBD; szczepy indica o wysokiej zawartości THC bez CBD; szczep z równą ilością CBD i THC oraz szczepy bogate w CBD. Co ciekawe, większość pacjentów otrzymywała więcej niż jeden szczep. Dziewięćset dwóch (24,9%) pacjentów zmarło, a 682 (18,8%) pacjentów przerwało leczenie po 6 miesiącach obserwacji. Spośród pozostałych pacjentów 60,6% odpowiedziało na leczenie; U 95,9% pacjentów nastąpiła znacząca poprawa stanu, u 3,7% — brak zmian, u 0,3% — pogorszenie. Przed rozpoczęciem leczenia tylko 18,7% pacjentów stwierdziło dobrą jakość życia, natomiast po 6 miesiącach leczenia —69,5%. Spośród wszystkich objawów związanych z rakiem najbardziej poprawiły się nudności, wymioty, depresja, migrena i zaburzenia snu. Najczęstszymi skutkami ubocznymi leczenia konopiami po 6 miesiącach obserwacji były zawroty głowy, suchość w jamie ustnej i zwiększony apetyt. Psychoaktywne działania niepożądane zauważyło tylko 2,8% pacjentów. Co ciekawe, spośród 344 pacjentów przyjmujących opioidy, 36% z nich przerwało ich przyjmowanie. Stwierdzono, że medyczna marihuana jest dobrze tolerowaną i bezpieczną opcją terapii paliatywnej dla pacjentów onkologicznych ( 109 ).
Rola konopi w wrodzonej i adaptacyjnej odpowiedzi immunologicznej
Będąc środkami immunomodulującymi, ekstrakty z konopi i pojedyncze kannabinoidy mogą wpływać zarówno na wrodzoną, jak i adaptacyjną odpowiedź immunologiczną. Ogólnie rzecz biorąc, kannabinoidy są uważane za związki immunosupresyjne. Wpływają na wrodzoną odpowiedź immunologiczną poprzez hamowanie aktywności komórek NK, komórek dendrytycznych, migrację neutrofili i makrofagów z ich procesami prezentacji antygenu i fagocytozy ( 110 ) oraz poprzez wyzwalanie indukcji MDSC ( 111 , 112 ). Zapalenie jest głównym mechanizmem wrodzonej odpowiedzi immunologicznej. Ogólnie rzecz biorąc, kannabinoidy, takie jak THC i CBD, powodują zmniejszenie ekspresji cytokin prozapalnych i zwiększenie ekspresji cytokin przeciwzapalnych. Dzięki temu aktywnie hamują proces zapalny ( 57 ). Jednak niektóre badania wykazują, że związki te mają różny wpływ na stan zapalny, albo go wzmacniając, albo tłumiąc. Na przykład CBD może aktywować odpowiedź immunologiczną poprzez podniesienie ekspresji mRNA TNF-α, IL-6, jak wykazano u myszy w odpowiedzi na zapalenie płuc wywołane LPS ( 113 ). Natomiast CBD hamowało IL-6 i IL-8 w mysim modelu raka jelita grubego in vivo opartym na linii komórkowej CT26 ( 104 ). Te sprzeczne wyniki mogą być specyficzne dla tkanki i dawki.
Kanabinoidy mogą wpływać na adaptacyjne odpowiedzi immunologiczne poprzez oddziaływanie na odporność humoralną i komórkową. Odporność komórek T może być pod wpływem kanabinoidów na różne sposoby: mogą one wpływać na proliferację i liczbę komórek T poprzez polaryzację odpowiedzi cytokinowej na Th1 lub Th2 ( 114 ). Wykazano, że kanabinoidy hamują proliferację komórek T, powodują ich apoptozę i wspierają polaryzację Th2 ( 115 , 116 ). Niektóre wstępne badania in vitro i in vivo THC wykazały immunosupresyjny wpływ na komórki T i komórki B przy stosowaniu wysokich stężeń, podczas gdy efekty immunostymulujące obserwowano przy niskich stężeniach ( 110 ). Badania eksperymentalne przeprowadzone in vivo na makakach zakażonych wirusem SIV, które otrzymywały THC przez okres 17 miesięcy, wykazały wzrost liczby komórek T, zmniejszenie ładunku wirusowego i wzrost ekspresji cytokin Th2 ( 117 ). Inne badanie przeprowadzone na pacjentach z HIV wykazało wyższe stężenie komórek CD4+ i CD8+ u pacjentów z THC-dodatnim wynikiem w porównaniu z pacjentami z THC-ujemnym wynikiem ( 118 ). Jeśli chodzi o rolę CBD, wykazano również, że może ono działać jako immunosupresant Th2 in vitro i in vivo , polaryzując odpowiedź cytokinową na Th2 i działając jako immunostymulant na Th1 ( 119 ). Jeśli chodzi o odporność humoralną, niektóre raporty z badań na ludziach wykazały zmniejszoną liczbę limfocytów B i zmniejszoną ilość IgM i IgG po spożyciu kannabinoidów w postaci bhang ( 120 ).
Przyszłe perspektywy wzmocnienia immunoterapii za pomocą kannabinoidów i ekstraktów z Cannabis Sativa
Immunomodulacyjne działanie konopi jest dobrze udokumentowane. Obecnie istnieje wiele znanych odmian konopi, a każda z nich ma unikalny skład różnych związków. Wiele badań wykazało wpływ pojedynczych kannabinoidów, takich jak THC i CBD, na stany zapalne i wzrost komórek rakowych ( 98 ). Inne składniki rośliny (takie jak mniejsze kannabinoidy, terpeny, terpenoidy, flawonoidy i inne) mogą działać synergicznie z kannabinoidami i mogą być przydatne z terapeutycznego punktu widzenia. Modulujący wpływ tych związków jest znany jako „efekt otoczenia”; taka modulacja jest zazwyczaj dodatnia, co oznacza, że efekt leczniczy całego ekstraktu z rośliny jest bardziej znaczący niż efekt izolowanych związków ( 121 ). Podobnie jak w przypadku każdego innego leku, efekty w znacznym stopniu zależą od stężenia. W przyszłości, dzięki dalszym badaniom, możemy uzyskać więcej informacji na temat potencjalnego immunostymulującego wpływu pojedynczych kannabinoidów lub ekstraktów z konopi. Wiedza ta może pomóc personelowi medycznemu w integracji ekstraktów z konopi z ukierunkowaną terapią nowotworową, potencjalnie jako terapia wspomagająca. Należy zidentyfikować specjalne ekstrakty o silnym działaniu przeciwnowotworowym, które nie są cytotoksyczne dla normalnych komórek i mogą uwrażliwiać komórki nowotworowe na dalsze leczenie bez zmniejszania odpowiedzi immunologicznej. Następnie te ekstrakty można połączyć z immunoterapią, a takie połączenie może mieć działanie synergistyczne. Wyniki retrospektywnej analizy przeprowadzonej u pacjentów z czerniakiem, rakiem nerki i niedrobnokomórkowym rakiem płuc, gdy konopie były stosowane w połączeniu z lekiem immunoterapeutycznym Nivolumab, wykazały zmniejszenie RR, ale nie zmiany w PFS i OS. Potrzebne są dalsze badania w celu zbadania możliwych interakcji między kannabinoidami a lekami immunoterapeutycznymi ( 122 ).
Należy przeprowadzić dogłębną eksplorację badań nad konopiami i powiązanymi lekami. Obecnie dysponujemy ograniczonymi danymi na temat interakcji konopi z innymi lekami, zwłaszcza w przypadku terapii ukierunkowanej. Ponieważ inhibitory punktów kontrolnych układu odpornościowego są rodzajem najbardziej skutecznej i efektywnej immunoterapii dla pacjentów z rakiem jelita grubego. Dlatego też należy przeprowadzić badania nad możliwością wzmocnienia immunoterapii za pomocą ekstraktów z konopi ( Rysunki 2 , 3 ).
Rysunek 2.
Potencjał kannabinoidów w immunoterapii nowotworów. Górny panel pokazuje, jak kannabinoidy mogą zwiększyć immunogenność guza. Uwalnianie antygenów nowotworowych może być zwiększone ze względu na bezpośrednią cytotoksyczność kannabinoidów w komórkach nowotworowych. Następnie następuje prezentacja wzmocnionych antygenów nowotworowych, po której następuje wzrost odpowiedzi immunologicznej zależnej od limfocytów T i liza komórek nowotworowych przez limfocyty T. Dolny panel pokazuje, jak kannabinoidy mogą odwrócić immunosupresję guza. Makrofagi można przeprogramować do fenotypu przeciwnowotworowego za pomocą kannabinoidów. Immunostymulujące makrofagi M1 wydzielają cytokiny przeciwnowotworowe i skutecznie fagocytują komórki nowotworowe.
Rysunek 3.
Pożądane efekty kannabinoidów na komórki odpornościowe.
Pierwsze podejście mogłoby się skupić na znalezieniu ekstraktów, które mogą zwiększyć immunogenność guza. Z tego powodu komórki nowotworowe będą bardziej podatne na rozpoznanie przez układ odpornościowy. W takim przypadku badania in vivo byłyby korzystne, aby zbadać konopie indyjskie jako terapię neoadjuwantową przed rozpoczęciem stosowania leków biologicznych. Powinny być one następnie poddane badaniom sprawdzającym, czy występuje wzmocnienie adaptacyjnych odpowiedzi immunologicznych pośredniczonych przez limfocyty T. Poprzez kierowanie cytotoksyczności do komórek rakowych, ekstrakty z konopi indyjskich mogą zwiększyć uwalnianie antygenów nowotworowych, a następnie wzmocnić prezentację antygenu. Należy również ocenić zmiany w mikrośrodowisku guza, takie jak naciek MDSC i Treg. Drugie podejście mogłoby się skupić na możliwości odwrócenia immunosupresji wywołanej przez guz za pomocą ekstraktów. Na przykład można to zrobić, przeprogramowując makrofagi na fenotyp przeciwnowotworowy. Będąc wysoce plastycznymi komórkami, makrofagi mogą łatwo przejść z typu pro-nowotworowego na typ przeciwnowotworowy. Niektóre badania in vivo wykazały, że wpływanie na szlak PI3kγ w makrofagach może dalej prowadzić do ich polaryzacji do typu immunostymulującego poprzez zwiększenie cytotoksyczności komórek CD8+ T i poprawę odpowiedzi na ICIs ( 123 ). Znajdując ekstrakty, które mogą wpływać na ten konkretny szlak, można połączyć je z immunoterapią, aby wykazać działanie synergistyczne. Ponadto ocenę polaryzacji makrofagów można przeprowadzić poprzez badanie cytokin. Ekstrakty z konopi, które polaryzują makrofagi do typu M1 i nie posiadają właściwości przeciwzapalnych, można dalej łączyć z immunoterapią.
Wniosek
W literaturze silnie sugerowano, że kannabinoidy i ekstrakty z konopi mogą być stosowane w leczeniu raka jelita grubego. Dowody wskazują, że kannabinoidy mają duży potencjał przekształcenia się w obiecujące leki. Oczywiste jest, że związki te mogą oddziaływać na kluczowe szlaki sygnałowe rozwoju raka. Ponadto należy przeprowadzić więcej przedklinicznych i klinicznych ocen kannabinoidów jako środków przeciw CRC i immunomodulujących. Dodatkowe badania pomogłyby nam znaleźć nowe możliwości zapobiegawcze i terapeutyczne dla pacjentów, u których istnieje ryzyko rozwoju CRC lub którzy obecnie się z nim zmagają. Wprowadzając te silne związki do obecnych protokołów leczenia, można osiągnąć redukcję dawki innych leków, które są wysoce toksyczne, zmniejszając w ten sposób niepożądane skutki uboczne; podobnie kannabinoidy mogą prawdopodobnie uwrażliwić komórki złośliwe na dalszą ukierunkowaną terapię.
Ponadto dalsze badania nowotworów, zwłaszcza ich sekwencjonowanie, dostarczą więcej informacji na temat ich specyficznych cech. Wiedza o tym, w jaki sposób ścieżki genetyczne oddziałują z pewnymi kannabinoidami, może pomóc lekarzom przepisywać je zgodnie z genetycznym składem nowotworu. Z tego powodu personel medyczny będzie przepisywał terapie oparte na kannabinoidach konkretnemu pacjentowi z konkretną chorobą nowotworową. Leczenie stanie się zorientowane na pacjenta i specyficzne dla wskazań. W rezultacie rokowanie w przypadku nowotworu i jego wskaźnik przeżycia mogą się znacznie poprawić.
W świetle szybko zmieniającego się krajobrazu dotyczącego legalizacji marihuany do celów medycznych i rekreacyjnych, pacjenci mogą być bardziej skłonni pytać lekarzy o jej potencjalne negatywne i korzystne skutki dla zdrowia. Popularne przekonanie wydaje się być takie, że marihuana jest nieszkodliwą przyjemnością, do której dostęp nie powinien być regulowany ani uważany za nielegalny. Obecnie marihuana jest najczęściej używanym „nielegalnym” narkotykiem w Stanach Zjednoczonych, przy czym około 12% osób w wieku 12 lat i starszych zgłosiło używanie w ciągu ostatniego roku, a szczególnie wysokie wskaźniki używania występują wśród osób młodych. 1 Najczęstszą drogą podawania jest inhalacja. Zielonkawo-szare poszarpane liście i kwiaty rośliny Cannabis sativa są palone (wraz z łodygami i nasionami) w papierosach, cygarach, fajkach, fajkach wodnych lub „bluntach” (marihuana zwinięta w tytoniowy liść z cygara). Haszysz jest pokrewnym produktem wytwarzanym z żywicy kwiatów marihuany i jest zwykle palony (sam lub w mieszance z tytoniem), ale może być przyjmowany doustnie. Marihuanę można wykorzystać także do zaparzania herbaty, a wyciąg z jej oleju można dodawać do produktów spożywczych.
Regularne używanie marihuany w okresie dojrzewania jest szczególnie niepokojące, ponieważ używanie jej przez tę grupę wiekową wiąże się ze zwiększonym prawdopodobieństwem wystąpienia szkodliwych skutków 2 ( Tabela 1 ). Chociaż wiele badań wykazało szkodliwe skutki, inne nie, a kwestia, czy marihuana jest szkodliwa, pozostaje przedmiotem gorącej debaty. W niniejszym artykule dokonujemy przeglądu obecnego stanu wiedzy naukowej na temat niekorzystnych skutków zdrowotnych rekreacyjnego używania marihuany, skupiając się na tych obszarach, w których dowody są najsilniejsze.
Tabela 1.
Skutki uboczne krótkotrwałego oraz długotrwałego i intensywnego stosowania marihuany.
Skutki krótkotrwałego stosowania
Osłabiona pamięć krótkotrwała, utrudniająca uczenie się i zapamiętywanie informacji
Zaburzenia koordynacji ruchowej, utrudniające prowadzenie pojazdu i zwiększające ryzyko obrażeń
Zmiana osądu, zwiększająca ryzyko zachowań seksualnych, które ułatwiają przenoszenie chorób przenoszonych drogą płciową
W dużych dawkach paranoja i psychoza
Skutki długotrwałego lub intensywnego stosowania
Uzależnienie (u ok. 9% użytkowników ogółem, 17% osób, które zaczynają używać w okresie dojrzewania i 25–50% osób, które używają codziennie) *
Słabe wyniki w nauce, zwiększone prawdopodobieństwo porzucenia szkoły *
Osłabienie funkcji poznawczych, z niższym IQ wśród osób, które często korzystały z narkotyków w okresie dojrzewania *
Zmniejszone zadowolenie z życia i osiągnięcia (określone na podstawie miar subiektywnych i obiektywnych w porównaniu z ocenami w populacji ogólnej) *
Objawy przewlekłego zapalenia oskrzeli
Zwiększone ryzyko przewlekłych zaburzeń psychotycznych (w tym schizofrenii) u osób ze skłonnością do takich zaburzeń
DZIAŁANIA NIEPOŻĄDANE
RYZYKO UZALEŻNIENIA
Pomimo pewnych kontrowersyjnych dyskusji na temat uzależniającego działania marihuany, dowody jasno wskazują, że długotrwałe używanie marihuany może prowadzić do uzależnienia. Rzeczywiście, około 9% osób, które eksperymentują z marihuaną, uzależni się 3 (zgodnie z kryteriami uzależnienia zawartymi w Diagnostic and Statistical Manual of Mental Disorders , 4th edition [DSM-IV]). Liczba ta wzrasta do około 1 na 6 wśród osób, które zaczynają używać marihuany jako nastolatki i do 25-50% wśród osób, które palą marihuanę codziennie. 4 Zgodnie z Narodowym Badaniem Używania Narkotyków i Zdrowia z 2012 r. szacunkowo 2,7 miliona osób w wieku 12 lat i starszych spełniło kryteria DSM-IV dotyczące uzależnienia od marihuany, a 5,1 miliona osób spełniło kryteria dotyczące uzależnienia od jakiegokolwiek nielegalnego narkotyku 1 (8,6 miliona spełniło kryteria dotyczące uzależnienia od alkoholu 1 ). Istnieje również rozpoznanie prawdziwego zespołu odstawienia marihuany 5 (obejmującego objawy takie jak drażliwość, trudności ze snem, dysforia, głód i lęk), który utrudnia zaprzestanie i przyczynia się do nawrotu. Używanie marihuany przez nastolatków jest szczególnie kłopotliwe. Zwiększona podatność nastolatków na niekorzystne długoterminowe skutki używania marihuany jest prawdopodobnie związana z faktem, że mózg, w tym układ endokannabinoidowy, przechodzi aktywny rozwój w okresie dojrzewania. 6 Rzeczywiście, wczesne i regularne używanie marihuany przewiduje zwiększone ryzyko uzależnienia od marihuany, co z kolei przewiduje zwiększone ryzyko używania innych nielegalnych narkotyków. 7 W porównaniu z osobami, które zaczynają używać marihuany w wieku dorosłym, osoby, które zaczynają w okresie dojrzewania, mają około 2 do 4 razy większe prawdopodobieństwo wystąpienia objawów uzależnienia od marihuany w ciągu 2 lat od pierwszego użycia. 8
WPŁYW NA ROZWÓJ MÓZGU
Mózg pozostaje w stanie aktywnego, kierowanego doświadczeniem rozwoju od okresu prenatalnego przez dzieciństwo i okres dojrzewania do wieku około 21 lat. 9 W tych okresach rozwojowych jest on z natury bardziej podatny niż mózg dojrzały na niekorzystne, długoterminowe skutki czynników środowiskowych, takich jak ekspozycja na tetrahydrokanabinol, lub THC, główny składnik aktywny marihuany. Pogląd ten uzyskał znaczne poparcie w badaniach na zwierzętach, które wykazały na przykład, że prenatalna lub młodzieńcza ekspozycja na THC może ponownie skalibrować wrażliwość układu nagrody na inne narkotyki 10 i że prenatalna ekspozycja zakłóca dynamikę cytoszkieletu, która jest krytyczna dla ustanowienia połączeń aksonalnych między neuronami. 11
W porównaniu z osobami nienarażonymi na działanie marihuany, dorośli, którzy regularnie palili marihuanę w okresie dojrzewania, mają upośledzoną łączność nerwową (mniej włókien) w określonych obszarach mózgu. Należą do nich przedklinek, kluczowy węzeł zaangażowany w funkcje wymagające wysokiego stopnia integracji (np. czujność i samoświadomość), a także strzępki, obszar hipokampa, który jest ważny w uczeniu się i zapamiętywaniu. 12 Zmniejszona łączność funkcjonalna została również zgłoszona w sieciach przedczołowych odpowiedzialnych za funkcje wykonawcze (w tym kontrolę hamującą) i sieciach podkorowych, które przetwarzają nawyki i rutyny. 13 Ponadto badania obrazowe u osób używających marihuany wykazały zmniejszoną aktywność w obszarach przedczołowych i zmniejszone objętości w hipokampie. 14 Tak więc niektóre obszary mózgu mogą być bardziej podatne niż inne na długotrwałe skutki marihuany. Jedno z badań wykazało, że selektywne zmniejszenie ekspresji receptorów kannabinoidowych-1 (CB1) w kilku obszarach kory mózgowej u długotrwałych palaczy marihuany było powiązane z wieloletnim paleniem konopi i ustępowało po 4 tygodniach abstynencji. 15 Zmian w receptorach CB1 nie zaobserwowano w obszarach podkorowych.
Dane epidemiologiczne i przedkliniczne sugerują, że używanie marihuany w okresie dojrzewania może wpływać na wiele zachowań uzależniających w wieku dorosłym. U gryzoni narażonych na kannabinoidy w okresie dojrzewania występuje zmniejszona reaktywność neuronów dopaminowych, które modulują obszary nagrody w mózgu. 18 Narażenie gryzoni na działanie konopi w macicy zmienia regulację rozwojową układu dopaminowego mezolimbicznego u dotkniętego tym problemem potomstwa. 19 Jeśli zmniejszona reaktywność dopaminy w obszarach nagrody w mózgu następuje po wczesnym narażeniu na działanie marihuany, efekt ten może pomóc wyjaśnić zwiększoną podatność na nadużywanie narkotyków i uzależnienie od kilku narkotyków w późniejszym życiu, o czym donoszono w większości badań epidemiologicznych. 20 Ta teoria jest również zgodna z modelami zwierzęcymi, które pokazują, że THC może przygotowywać mózg do wzmocnionych reakcji na inne narkotyki. 21 Chociaż wyniki te potwierdzają tezę, że marihuana jest narkotykiem wprowadzającym, inne narkotyki, takie jak alkohol i nikotyna, również można zaliczyć do tej kategorii, ponieważ one również przygotowują mózg na wzmożoną reakcję na inne narkotyki. 22 Jednak alternatywne wyjaśnienie jest takie, że osoby, które są bardziej podatne na zachowania związane z przyjmowaniem narkotyków, po prostu częściej zaczynają od marihuany ze względu na jej dostępność, a ich późniejsze interakcje społeczne z innymi użytkownikami narkotyków zwiększają prawdopodobieństwo, że spróbują innych narkotyków.
ZWIĄZEK Z CHOROBĄ PSYCHICZNĄ
Regularne używanie marihuany wiąże się ze zwiększonym ryzykiem lęku i depresji, 23 ale związek przyczynowo-skutkowy nie został ustalony. Marihuana jest również powiązana z psychozami (w tym tymi związanymi ze schizofrenią), szczególnie u osób z istniejącą wcześniej podatnością genetyczną, 24 i zaostrza przebieg choroby u pacjentów ze schizofrenią. Intensywniejsze używanie marihuany, większa moc leku i narażenie w młodszym wieku mogą negatywnie wpłynąć na przebieg choroby (np. poprzez przyspieszenie czasu pierwszego epizodu psychotycznego o 2 do 6 lat). 25
Jednakże w tego typu badaniach trudno jest ustalić związek przyczynowo-skutkowy, ponieważ czynniki inne niż używanie marihuany mogą być bezpośrednio związane z ryzykiem choroby psychicznej. Ponadto inne czynniki mogą predysponować osobę zarówno do używania marihuany, jak i choroby psychicznej. Utrudnia to pewne przypisanie zwiększonego ryzyka choroby psychicznej używaniu marihuany.
WPŁYW NA WYNIKI SZKOLNE I OSIĄGNIĘCIA W CAŁYM ŻYCIU
W badaniu Monitoring the Future z 2013 r. wśród uczniów szkół średnich, 26 6,5% uczniów w klasie 12 zgłosiło codzienne lub prawie codzienne używanie marihuany, a liczba ta prawdopodobnie stanowi niedoszacowanie używania, ponieważ młodzi ludzie, którzy porzucili szkołę, mogą mieć szczególnie wysokie wskaźniki częstego używania marihuany. 27 Ponieważ używanie marihuany upośledza kluczowe funkcje poznawcze, zarówno podczas ostrego odurzenia, jak i przez kilka dni po użyciu, 28 wielu uczniów może funkcjonować na poziomie poznawczym, który jest poniżej ich naturalnych możliwości przez znaczne okresy czasu. Chociaż ostre efekty mogą ustąpić po usunięciu THC z mózgu, mimo to stwarza poważne ryzyko dla zdrowia, którego można się spodziewać przy długotrwałym lub intensywnym używaniu. Dowody sugerują, że takie używanie skutkuje mierzalnymi i długotrwałymi upośledzeniami poznawczymi, 16 szczególnie wśród tych, którzy zaczęli używać marihuany we wczesnej adolescencji. Co więcej, niezdolność do nauki w szkole, nawet przez krótkie lub sporadyczne okresy (wtórny skutek ostrego zatrucia), będzie miała wpływ na późniejszą zdolność do osiągania coraz trudniejszych celów edukacyjnych, co może również wyjaśniać związek między regularnym używaniem marihuany a słabymi ocenami.29
Związek między używaniem marihuany przez młodych ludzi a szkodami psychospołecznymi jest prawdopodobnie wieloaspektowy, co może wyjaśniać niespójności między badaniami. Na przykład, niektóre badania sugerują, że długoterminowe deficyty mogą być odwracalne i pozostać subtelne, a nie upośledzające, gdy dana osoba powstrzyma się od używania. 30 Inne badania pokazują, że długotrwałe, intensywne używanie marihuany powoduje upośledzenie pamięci i uwagi, które utrzymuje się i pogarsza wraz z kolejnymi latami regularnego używania31 i wraz z rozpoczęciem używania w okresie dojrzewania. 32 Jak zauważono powyżej, wczesne używanie marihuany wiąże się z pogorszeniem wyników w szkole i zwiększonym ryzykiem porzucenia szkoły, 27 , 29 chociaż doniesienia o wspólnych czynnikach środowiskowych, które wpływają na ryzyko używania marihuany w młodym wieku i porzucenia szkoły33 sugerują , że związek ten może być bardziej złożony. Intensywne używanie marihuany wiąże się z niższymi dochodami, większym zapotrzebowaniem na pomoc społeczno-ekonomiczną, bezrobociem, zachowaniami przestępczymi i niższym zadowoleniem z życia. 2 , 34
RYZYKO WYPADKÓW SAMOCHODOWYCH
Zarówno natychmiastowa, jak i długotrwała ekspozycja na marihuanę upośledzają zdolność prowadzenia pojazdów; marihuana jest nielegalnym narkotykiem najczęściej zgłaszanym w związku z upośledzoną jazdą i wypadkami, w tym wypadkami śmiertelnymi. 35 Istnieje związek między stężeniem THC we krwi a wynikami w kontrolowanych badaniach symulujących jazdę, 36 które są dobrym wskaźnikiem rzeczywistej zdolności prowadzenia pojazdów. Niedawne palenie marihuany i poziom THC we krwi wynoszący od 2 do 5 ng na mililitr są związane ze znacznym upośledzeniem zdolności prowadzenia pojazdów. 37 Zgodnie z metaanalizą, ogólne ryzyko udziału w wypadku wzrasta o współczynnik około 2, gdy osoba prowadzi pojazd wkrótce po użyciu marihuany. 37 W analizie winy za wypadek osoby z pozytywnym wynikiem testu na obecność THC (typowy minimalny poziom wykrywalności, 1 ng na mililitr), a w szczególności osoby z wyższym poziomem we krwi, były 3 do 7 razy bardziej narażone na spowodowanie wypadku samochodowego niż osoby, które nie używały narkotyków ani alkoholu przed prowadzeniem pojazdu. 38 Dla porównania, ogólne ryzyko wypadku samochodowego wzrasta prawie 5-krotnie w przypadku kierowców, u których stężenie alkoholu we krwi przekracza 0,08%, czyli dopuszczalny limit w większości krajów, a w przypadku osób poniżej 21. roku życia wzrasta 27-krotnie. 39 Nic dziwnego, że ryzyko związane ze stosowaniem alkoholu w połączeniu z marihuaną wydaje się być większe niż ryzyko związane ze stosowaniem każdego z tych narkotyków osobno. 37
RYZYKO RAKA I INNE SKUTKI DLA ZDROWIA
Wpływ długotrwałego palenia marihuany na ryzyko raka płuc jest niejasny. Na przykład używanie marihuany przez okres równoważny 30 lub więcej latom (przy czym 1 rok używania marihuany jest równy 1 papierosowi [skrętowi] marihuany wypalanemu dziennie przez 1 rok) wiązało się ze zwiększoną częstością występowania raka płuc i kilku nowotworów górnego odcinka przewodu pokarmowego; jednak związek ten zanikł po uwzględnieniu potencjalnych czynników zakłócających, takich jak palenie papierosów. 40 Chociaż nie można wykluczyć możliwości pozytywnego związku między paleniem marihuany a rakiem, 41 dowody sugerują, że ryzyko jest niższe w przypadku marihuany niż tytoniu. 40 Jednak palenie papierosów zawierających zarówno marihuanę, jak i produkty tytoniowe jest potencjalnym czynnikiem zakłócającym, którego rozpowszechnienie znacznie różni się w zależności od kraju.
Palenie marihuany wiąże się również ze stanem zapalnym dużych dróg oddechowych, zwiększonym oporem w drogach oddechowych i hiperinflacją płuc, co jest zgodne z faktem, że regularni palacze marihuany częściej zgłaszają objawy przewlekłego zapalenia oskrzeli niż osoby niepalące42 ; jednak długoterminowy wpływ niewielkich dawek marihuany nie wydaje się być znaczący.43 Kompetencja immunologiczna układu oddechowego u palaczy marihuany może być również upośledzona, o czym świadczy zwiększona częstość infekcji dróg oddechowych i zapalenia płuc.44 Używanie marihuany wiąże się również z chorobami naczyń, które zwiększają ryzyko zawału mięśnia sercowego, udaru i przemijających ataków niedokrwiennych podczas zatrucia marihuaną.45 Rzeczywiste mechanizmy leżące u podstaw wpływu marihuany na układ sercowo-naczyniowy i mózgowo-naczyniowy są złożone i nie do końca poznane. Jednakże bezpośrednie działanie kannabinoidów na różne receptory docelowe (np. receptory CB1 w tętniczych naczyniach krwionośnych) i pośrednie działanie na związki wazoaktywne46 może pomóc wyjaśnić szkodliwy wpływ marihuany na opór naczyniowy i mikrokrążenie wieńcowe.47
OGRANICZENIA DOWODÓW I LUKI W WIEDZY
Większość długoterminowych skutków używania marihuany, które są tutaj podsumowane, zaobserwowano u użytkowników intensywnie lub długoterminowo, ale liczne (często ukryte) czynniki zakłócające utrudniają nam ustalenie związku przyczynowo-skutkowego (w tym częste używanie marihuany w połączeniu z innymi narkotykami). Czynniki te utrudniają również ocenę prawdziwego wpływu wewnątrzmacicznej ekspozycji na marihuanę. Rzeczywiście, pomimo używania marihuany przez kobiety w ciąży, 48 i modeli zwierzęcych sugerujących, że ekspozycja na konopie w czasie ciąży może zmieniać normalne procesy i trajektorie rozwoju mózgu, 49 nasze zrozumienie długoterminowych skutków prenatalnej ekspozycji na marihuanę u ludzi jest bardzo słabe.
Zawartość THC, czyli moc marihuany, wykryta w skonfiskowanych próbkach, stale wzrastała z około 3% w latach 80. do 12% w 2012 r. 50 ( rys. 1A ). Ten wzrost zawartości THC budzi obawy, że konsekwencje używania marihuany mogą być teraz gorsze niż w przeszłości i może wyjaśniać znaczny wzrost liczby wizyt na oddziałach ratunkowych osób zgłaszających używanie marihuany 51 ( rys. 1B ) i wzrost liczby śmiertelnych wypadków samochodowych. 35 Ten wzrost mocy THC w czasie budzi również pytania o obecną trafność ustaleń starszych badań nad skutkami używania marihuany, zwłaszcza badań oceniających długoterminowe wyniki.
Rysunek 1. Wzrost stężenia tetrahydrokanabinolu (THC) w marihuanie w czasie oraz liczba wizyt na oddziale ratunkowym w związku z marihuaną, kokainą lub heroiną.
Wykres A przedstawia wzrastającą moc marihuany (tj. procentową zawartość THC) w próbkach skonfiskowanych przez Drug Enforcement Administration (DEA) w latach 1995–2012. Wykres B przedstawia szacunki liczby wizyt na oddziałach ratunkowych związanych z zażywaniem wybranych narkotyków (marihuany, kokainy i heroiny) pojedynczo lub w połączeniu z innymi narkotykami w latach 2004–2011. Spośród tych trzech narkotyków jedynie marihuana, stosowana w połączeniu z innymi narkotykami lub samodzielnie, wiązała się ze znacznym wzrostem liczby wizyt w tym okresie (wzrost o 62% w przypadku stosowania w połączeniu z innymi narkotykami i wzrost o 100% w przypadku stosowania samodzielnie, P<0,05 dla obu porównań).
Istnieje również potrzeba lepszego zrozumienia, w jaki sposób wykorzystać potencjalne korzyści medyczne rośliny marihuany bez narażania osób chorych na jej wewnętrzne ryzyko. Autorytatywny raport Instytutu Medycyny, Marihuany i Medycyny52 uznaje potencjalne korzyści palenia marihuany w pobudzaniu apetytu, szczególnie u pacjentów z zespołem nabytego niedoboru odporności (AIDS) i powiązanym zespołem wyniszczenia, a także w zwalczaniu nudności i wymiotów wywołanych chemioterapią , silnego bólu i niektórych form spastyczności. Raport wskazuje również, że istnieją pewne dowody na korzyści ze stosowania marihuany w celu zmniejszenia ciśnienia śródgałkowego w leczeniu jaskry. Niemniej jednak raport podkreśla znaczenie skupienia wysiłków badawczych na terapeutycznym potencjale syntetycznych lub farmaceutycznie czystych kannabinoidów.52 Niektórzy lekarze nadal przepisują marihuanę w celach leczniczych, pomimo ograniczonych dowodów na korzyści (patrz ramka ). Praktyka ta budzi szczególne obawy w odniesieniu do długotrwałego stosowania przez wrażliwe populacje . Na przykład istnieją pewne dowody sugerujące, że u pacjentów z objawami zakażenia wirusem niedoboru odporności ludzkiej (HIV) lub AIDS, używanie marihuany może w rzeczywistości zaostrzać deficyty poznawcze związane z HIV. 75 Podobnie, potrzeba więcej badań, aby zrozumieć potencjalne skutki używania marihuany na związane z wiekiem pogorszenie funkcji poznawczych w ogóle, a w szczególności na upośledzenie pamięci.
Stany kliniczne z objawami, które można złagodzić leczeniem marihuaną lub innymi kannabinoidami.*.
Jaskra
Wczesne dowody korzyści marihuany u pacjentów z jaskrą (chorobą związaną ze zwiększonym ciśnieniem w oku) mogą być zgodne z jej zdolnością do wywoływania przejściowego obniżenia ciśnienia śródgałkowego, 53 , 54 ale inne, standardowe metody leczenia są obecnie bardziej skuteczne. Wykazano, że THC, kannabinol i nabilon (syntetyczny kannabinoid podobny do THC), ale nie kannabidiol, obniżają ciśnienie śródgałkowe u królików. 55 , 56 Potrzebne są dalsze badania, aby ustalić, czy cząsteczki, które modulują układ endokannabinoidowy, mogą nie tylko obniżać ciśnienie śródgałkowe, ale także zapewniać korzyści neuroprotekcyjne u pacjentów z jaskrą. 57
Mdłości
Leczenie nudności i wymiotów związanych z chemioterapią było jednym z pierwszych zastosowań medycznych THC i innych kannabinoidów. 58 THC jest skutecznym środkiem przeciwwymiotnym u pacjentów poddawanych chemioterapii, 59 ale pacjenci często twierdzą, że marihuana jest skuteczniejsza w tłumieniu nudności. Inne, niezidentyfikowane związki w marihuanie mogą wzmacniać działanie THC (jak to się wydaje w przypadku THC i kannabidiolu, które działają poprzez różne mechanizmy przeciwwymiotne). 60 Paradoksalnie, zwiększone wymioty (hiperemesis) zgłaszano w przypadku powtarzającego się stosowania marihuany.
Anoreksja i zespół wyniszczenia związany z AIDS
Doniesienia wskazują, że palenie lub spożywanie konopi poprawia apetyt i prowadzi do zwiększenia masy ciała oraz poprawy nastroju i jakości życia u pacjentów z AIDS. 61 Jednakże nie ma długoterminowych lub rygorystycznych dowodów na trwały wpływ konopi na zachorowalność i śmiertelność związaną z AIDS, przy akceptowalnym profilu bezpieczeństwa, który uzasadniałby jej włączenie do obecnej praktyki klinicznej u pacjentów otrzymujących skuteczną terapię antyretrowirusową. 62 Dane z nielicznych badań, które badały potencjalną wartość terapeutyczną kannabinoidów dla tej populacji pacjentów, są niejednoznaczne. 62
Przewlekły ból
Marihuana była stosowana w celu łagodzenia bólu od stuleci. Badania wykazały, że kannabinoidy działające poprzez centralne receptory CB1, a prawdopodobnie także obwodowe receptory CB1 i CB2, 63 odgrywają ważną rolę w modelowaniu odpowiedzi nocyceptywnych w różnych modelach bólu. Wyniki te są zgodne z doniesieniami, że marihuana może być skuteczna w łagodzeniu bólu neuropatycznego, 64 , 65 nawet przy bardzo niskich poziomach THC (1,29%). 66 Zarówno marihuana, jak i dronabinol, farmaceutyczna formuła THC, zmniejszają ból, ale dronabinol może prowadzić do dłużej trwającego zmniejszenia wrażliwości na ból i niższych ocen efektów nagradzających. 67
Zapalenie
Kanabinoidy (np. THC i kannabidiol) mają znaczące działanie przeciwzapalne ze względu na zdolność do wywoływania apoptozy, hamowania proliferacji komórek i tłumienia produkcji cytokin. 68 Kanabidiol wzbudził szczególne zainteresowanie jako środek przeciwzapalny ze względu na brak efektów psychoaktywnych. 58 Modele zwierzęce wykazały, że kannabidiol jest obiecującym kandydatem do leczenia reumatoidalnego zapalenia stawów 58 i chorób zapalnych przewodu pokarmowego (np. wrzodziejącego zapalenia jelita grubego i choroby Leśniowskiego-Crohna). 69
Stwardnienie rozsiane
Nabiximols (Sativex, GW Pharmaceuticals), oromucosalny spray, który dostarcza mieszankę THC i kannabidiolu, wydaje się być skutecznym leczeniem bólu neuropatycznego, zaburzeń snu i spastyczności u pacjentów ze stwardnieniem rozsianym. Sativex jest dostępny w Wielkiej Brytanii, Kanadzie i kilku innych krajach 70 , 71 i jest obecnie poddawany przeglądowi w badaniach fazy 3 w Stanach Zjednoczonych w celu uzyskania zatwierdzenia od Food and Drug Administration.
Padaczka
W niedawnym małym badaniu rodziców, którzy stosują marihuanę o wysokiej zawartości kannabidiolu w leczeniu napadów padaczkowych u swoich dzieci, 72 11% (2 rodziny z 19, które spełniły kryteria włączenia) zgłosiło całkowity brak napadów, 42% (8 rodzin) zgłosiło zmniejszenie częstości napadów o ponad 80%, a 32% (6 rodzin) zgłosiło zmniejszenie częstości napadów o 25 do 60%. Chociaż takie raporty są obiecujące, nie ma wystarczających danych dotyczących bezpieczeństwa i skuteczności stosowania roślin konopi w leczeniu padaczki. 73 Jednak coraz więcej dowodów wskazuje na rolę kannabidiolu jako środka przeciwpadaczkowego w modelach zwierzęcych. 74
*AIDS oznacza zespół nabytego niedoboru odporności, receptor kannabinoidowy-1 CB1 i receptor kannabinoidowy-2 CB2, wirus niedoboru odporności ludzkiej HIV i tetrahydrokanabinol THC.
Potrzebne są badania nad sposobami, w jakie polityka rządowa dotycząca marihuany wpływa na wyniki w zakresie zdrowia publicznego. Nasza wiedza na temat wpływu polityki na siły rynkowe jest dość ograniczona (np. atrakcyjność nowych źródeł dochodów podatkowych z legalnej sprzedaży marihuany, wojny cenowe, reklamy skierowane do młodzieży i pojawienie się leków na bazie konopi zatwierdzonych przez Food and Drug Administration), podobnie jak nasza wiedza na temat powiązanych zmiennych postrzegania stosowania, rodzajów stosowania i wyników. Historycznie rzecz biorąc, istniała odwrotna korelacja między stosowaniem marihuany a postrzeganiem jej ryzyka wśród nastolatków ( rys. 2A ). Zakładając, że ta odwrotna zależność jest przyczynowa, czy większa pobłażliwość w kulturze i polityce społecznej doprowadziłaby do wzrostu liczby młodych ludzi, którzy są regularnie narażeni na działanie konopi? Wśród uczniów w klasie 12. zgłaszana częstość regularnego palenia marihuany stale rośnie w ostatnich latach i może wkrótce przeciąć linię trendu dla regularnego palenia tytoniu ( rys. 2B ). Potrzebujemy również informacji o skutkach biernego narażenia na dym z konopi i kannabinoidy. Bierne narażenie jest ważnym problemem zdrowia publicznego w kontekście palenia tytoniu, ale nie mamy jasnego zrozumienia skutków biernego narażenia na palenie marihuany. 76 Badania w stanach (np. Kolorado, Kalifornia i Waszyngton) i krajach (np. Urugwaj, Portugalia i Holandia), w których polityka społeczna i prawna ulega zmianie, mogą dostarczyć ważnych danych do kształtowania przyszłych polityk.
Rysunek 2. Używanie marihuany w odniesieniu do postrzeganego ryzyka i codziennego używania papierosów tytoniowych lub marihuany wśród uczniów amerykańskich klas 12. w latach 1975–2013.
Panel A pokazuje odwrotną korelację między postrzeganiem ryzyka związanego z używaniem marihuany a rzeczywistym używaniem. Postrzegane ryzyko odpowiada odsetkowi nastolatków, którzy zgłosili, że używanie marihuany jest niebezpieczne. Panel B pokazuje odsetek uczniów, którzy zgłosili codzienne używanie papierosów tytoniowych lub marihuany w ciągu ostatnich 30 dni. Dane dla obu wykresów pochodzą z Johnston et al. 26
WNIOSKI
Używanie marihuany wiąże się ze znacznymi skutkami ubocznymi, z których niektóre zostały określone z dużym prawdopodobieństwem ( Tabela 2 ). Marihuana, podobnie jak inne narkotyki, może powodować uzależnienie. Podczas odurzenia marihuana może zakłócać funkcje poznawcze (np. pamięć i postrzeganie czasu) i funkcje motoryczne (np. koordynację), a te efekty mogą mieć szkodliwe konsekwencje (np. wypadki samochodowe). Powtarzające się używanie marihuany w okresie dojrzewania może skutkować długotrwałymi zmianami w funkcjonowaniu mózgu, które mogą zagrozić osiągnięciom edukacyjnym, zawodowym i społecznym. Jednak skutki narkotyku (legalnego lub nielegalnego) na zdrowie jednostki są określane nie tylko przez jego właściwości farmakologiczne, ale także przez jego dostępność i akceptowalność społeczną. Pod tym względem legalne narkotyki (alkohol i tytoń) oferują trzeźwiącą perspektywę, odpowiadając za największe obciążenie chorobami związanymi z narkotykami 77 nie dlatego, że są bardziej niebezpieczne niż narkotyki nielegalne, ale dlatego, że ich status prawny pozwala na bardziej powszechną ekspozycję. W miarę jak polityka zmierza w kierunku legalizacji marihuany, rozsądne, a prawdopodobnie ostrożne, jest założenie, że wzrośnie jej użycie, a co za tym idzie, wzrośnie również liczba osób, u których wystąpią negatywne skutki zdrowotne.
Tabela 2.
Poziom zaufania do dowodów na negatywny wpływ marihuany na zdrowie i samopoczucie.
* Wskazany ogólny poziom pewności co do związku między używaniem marihuany a wymienionymi skutkami stanowi próbę oceny siły obecnych dowodów, zwłaszcza w odniesieniu do intensywnego lub długotrwałego używania oraz używania rozpoczynającego się w okresie dojrzewania.
Nierównowaga jelitowa prowadzi do zapalnej choroby jelit (IBD), a przewlekły stan zapalny predysponuje do onkogenezy. Komórki dendrytyczne prezentujące antygen (DC) i makrofagi mogą przechylić równowagę w kierunku tolerancji lub patologii. Tutaj pokazujemy, że delta-9-tetrahydrokannabinol (THC) osłabia raka jelita grubego związanego z zapaleniem jelita grubego i zapalenie jelita grubego wywołane przez przeciwciała anty-CD40. Działając poprzez receptor kannabinoidowy 2 (CB2), THC zwiększa ekspresję CD103 na DC i makrofagach oraz zwiększa ekspresję TGF-β1, aby zwiększyć liczbę komórek regulatorowych T (Tregs). Komórki Tregs indukowane przez THC są niezbędne do zaradzenia systemicznemu IFNγ i TNFα wywołanemu przez przeciwciała anty-CD40, ale hamowanie APC przez CB2 przez THC gasi patogenne uwalnianie IL-22 i IL-17A w jelicie grubym. Badając tkanki z wielu miejsc, potwierdziliśmy, że THC wpływa na DC, szczególnie w miejscach bariery śluzowej w okrężnicy i płucach, aby zmniejszyć DC CD86. Używając modeli zapalenia okrężnicy i zapalenia układowego wykazujemy, że THC, poprzez CB2, jest silnym supresorem nieprawidłowych odpowiedzi immunologicznych, prowokując koordynację między APC i Treg.
THC może zapobiegać rozwojowi raka jelita grubego u myszy wywołanego zapaleniem okrężnicy
•Aktywacja CB2 w komórkach układu odpornościowego tłumi chorobotwórczą onkogenną IL-22
•THC osłabia zapalenie jelit wywołane przez anty-CD40 poprzez Tregs i APCs
•Aktywacja CB2 na komórkach APC przez THC zmniejsza wydzielanie cytokin prozapalnych
Równowaga między tolerancją immunologiczną a uprzedzeniami w jelitach wymaga precyzyjnie dostrojonych sygnałów molekularnych, aby zapobiec wadom, które mogą prowadzić do chorób zapalnych jelit (IBD), choroby Leśniowskiego-Crohna i wrzodziejącego zapalenia jelita grubego ( Lynch i Pedersen, 2016 ). Stany te charakteryzują się nieograniczonym zapaleniem przewodu pokarmowego, które powoduje zniszczenie bariery śluzówkowej. Obciążenie IBD rośnie na całym świecie, a zapadalność i rozpowszechnienie zwiększają się szczególnie w krajach wschodnich i rozwijających się ( Coward i in., 2019 ). Wykazano jednoznacznie, że oprócz zgubnych skutków IBD osoby dotknięte tą chorobą mają zwiększone ryzyko zachorowania na raka jelita grubego (CRC) w ciągu swojego życia ( Dyson i Rutter, 2012 ). Chociaż pełna etiologia IBD jest niejasna, u podstaw jej rozwoju leży wyraźny składnik genetyczny ( Cho, 2008 ), a ostatnie badania dokładnie zmapowały 45 potencjalnych loci jako wyzwalające IBD ( Huang i in., 2017 ). Pomimo postępu w określeniu genetycznej podatności na IBD, czynniki genetyczne żywiciela nie mogą odpowiadać za rosnącą zapadalność na tę chorobę na całym świecie. Dlatego należy brać pod uwagę czynniki środowiskowe i mikrobiologiczne, a także odpowiedź immunologiczną na nie.
Główne geny podatności na IBD, w tym NOD2, STAT3 i IL-23 ( Cho, 2008 ; Huang i in., 2017 ; Duerr i in., 2006 ; de Souza i Fiocchi, 2016 ), są silnie zaangażowane w procesy immunologiczne. W związku z tym stężenia interleukiny (IL)-23, IL-17 i IL-22 występują na wyższych poziomach w surowicy i jelitach pacjentów z IBD ( Brand i in., 2006 ; Fujino i in., 2003 ). Prozapalne IL-17 i IL-23 odgrywają dobrze zdefiniowaną rolę w kaskadach zapalnych, które wywołują IBD; jednak rola IL-22 jest kwestią sporną. IL-22 jest kluczowa w regulacji powierzchni barierowych: zwiększaniu obrony nabłonkowej i śluzówkowej, uwalnianiu peptydów przeciwdrobnoustrojowych ( Parks i in., 2016 ) i ochronie komórek macierzystych enterocytów ( Gronke i in., 2019 ) i jest niezbędna do usunięcia zakażenia Citrobacter rodentium ( Manta i in., 2013 ). IL-22 została również zidentyfikowana jako patogenna ( Eken i in., 2014 ), a IL-22 jest silnym kandydatem do inicjowania CRC poprzez zwiększanie ekspresji STAT3 w komórkach nabłonkowych ( Sun i in., 2015 ; Kryczek i in., 2014 ; Kirchberger i in., 2013 ). Głównymi producentami IL-22 w jelitach są wrodzone komórki limfoidalne grupy 3 (ILC3) i komórki Th CD4+, które wydzielają IL-22 na niższych poziomach ( Dudakov i in., 2015 ). Produkcja IL-22 zarówno przez komórki ILC3, jak i Th22 może być stymulowana przez prozapalne cytokiny IL-23, MCP-1, IL-1β i IL-6 ( Parks i in., 2016 ; Manta i in., 2013 ; Dudakov i in., 2015 ). Produkcja IL-22 przez ILC3 jest ściśle kontrolowana przez czynniki egzogenne i endogenne. Metabolity z pożywienia i drobnoustrojów modulują czynniki transkrypcyjne, takie jak AhR, RORγt i STAT3 ( Kiss i Diefenbach, 2012 ). Endogenna aktywność komórkowa pomiędzy ILC3 a komórkami prezentującymi antygen (APC) może również regulować wydzielanie ILC3 IL-22. Komórki APC w jelitach są arbitrami pomiędzy tolerancją a opornością, przygotowując ILC, komórki Th i chroniące jelita komórki regulatorowe T (Tregs) przed uznanymi zagrożeniami. Komórki dendrytyczne (DC) są klasycznymi komórkami APC i prezentują antygeny komensalne i dietetyczne komórkom T w jelitach. Komórki DC w jelitach nie są jednorodne. Komórki DC CD103+ są zazwyczaj tolerogenne, wyrażając wysokie poziomy dehydrogenazy aldehydowej, TGF-β1 i integryny β8, które działają w celu indukowania komórek Tregs i regulacji w górę CCR9 w celu skierowania się do jelit ( Sun i in., 2007 ; Merad i in., 2013 ; Coombes i in., 2007). Uważa się jednak, że zwiększona ekspresja CD103 na DC odgrywa rolę w krzyżowym inicjowaniu wirusa i cytotoksycznych komórek T specyficznych dla nowotworów ( Hildner i in., 2008 ), a rola CD103+ CD11b+ DC jest mniej jasna, wykazując fenotyp miecza obosiecznego, w którym mogą zachowywać się jak CD103+ DC, wydzielając kwas retinowy i zwiększając liczbę Treg, lub mogą indukować komórki Th17 ( Persson i in., 2013 ). CD103- CD11b+ DC działają w bardziej klasyczny sposób prozapalny, podobnie jak makrofagi. Makrofagi w jelitach, zwłaszcza tkankowo rezydujące makrofagi CX 3 CR1+ (receptor fraktalkiny) (zwane również komórkami dendrytycznymi mieloidalnymi lub fagocytami jednojądrowymi) są dominującym źródłem cytokin prozapalnych, IL-23, IL-1β i czynnika stymulującego kolonie granulocytów i makrofagów (GM-CSF) w jelitach, które są silnymi aktywatorami produkcji ILC3 IL-22, a usunięcie tych komórek zmniejsza odsetek ILC3 wytwarzających IL-22 ( Manta i in., 2013 ; Longman i in., 2014 ).
Modulacja układu endokannabinoidowego (ECS) w przypadku IBD okazała się obiecująca w modelach przedklinicznych ( Whiting i in., 2015 ; Singh i in., 2012 ; Storr i in., 2009 ; Ke i in., 2016 ) i małych kohortach ludzkich ( Naftali i in., 2013 ; Lahat i in., 2012 ). Dane mysie dotyczące ECS w CRC wskazują na rolę kannabinoidów w zapobieganiu nieprawidłowemu tworzeniu krypt przez CB2 ( Izzo i in., 2008 ) i bezpośredniej apoptozie komórek nowotworowych przez CB1 lub CB2 ( Cianchi i in., 2008 ). Dane ludzkie wskazują, że CB2 jest nadregulowane w tkance guza jelita grubego, a guzy wyrażające wyższy poziom mRNA CB2 bardziej proliferują ( Martínez-Martínez i in., 2015 ). Rozbieżność między obserwacjami aktywności ECS w raku można częściowo wyjaśnić ustaleniami, że agoniści receptora kannabinoidowego (CB) wykazują bimodalne działanie w liniach komórek rakowych z odpowiedziami proliferacyjnymi przy niższych poziomach endogennych i odpowiedziami apoptotycznymi przy wyższych dawkach egzogennych ( Pisanti i in., 2013 ). W tym badaniu wykorzystujemy egzogenny kannabinoid delta-9-tetrahydrokannabinol (THC) w celu wpłynięcia na równowagę immunologiczną jelit w kierunku homeostazy w modelach stanu zapalnego nowotworowego i przednowotworowego. Wykazaliśmy, że THC jest skutecznym leczeniem w zapobieganiu rakowi jelita grubego związanemu z zapaleniem okrężnicy, wykorzystując model azoksymetanu (AOM) + siarczanu sodu dekstranu (DSS) w nowotworzeniu jelita grubego. Wykazaliśmy również, że pronowotworowa IL-22 w mikrośrodowisku nabłonka jest redukowana poprzez aktywację CB2 zależną od THC w komórkach hematopoetycznych. Aby wyizolować wpływ THC na układ odpornościowy i zbadać interakcje między odpornością adaptacyjną jelit a wrodzoną w celu regulacji wydzielania IL-22, zastosowaliśmy model anty-CD40, który inicjuje przebieg choroby niezależnie od mikrobioty lub przepuszczalności jelit w celu stymulacji stanu zapalnego ( Uhlig i in., 2006 ; Barthels i in., 2017 ). Interakcja CD40-CD154 (CD40L) między APC i limfocytami T jest ważna dla inicjacji i utrzymania adaptacyjnych odpowiedzi immunologicznych ( Cong i in., 2000 ). Blokowanie interakcji CD40-CD40L może być stosowane profilaktycznie i terapeutycznie w ustalonych modelach zapalenia jelita grubego u myszy ( Polese i in., 2002 ). U ludzi, komórki CD40+ APC znajdują się w pobliżu limfocytów T CD40L+ w zapalonej tkance IBD ( De Jong i in., 2000 ). Badania wykazały, że stymulacja CD40 u myszy inicjuje kaskadę zapalną zależną od produkcji IL-12 przez komórki mieloidalne w celu wywołania uwalniania interferonu (IFN)-γ przez limfocyty T i komórki NK, co powoduje stan zapalny układowy, oraz uwalnianie IL-23 przez mieloidalne prowadzące do zapalenia okrężnicy ( Uhlig i in., 2006). U myszy z nienaruszoną odpornością adaptacyjną, Tregs odgrywają ważną rolę w rozwoju i odporności na choroby. LAG-3+ Tregs mogą zmniejszyć kolitogenne stymulowane CD40 wydzielanie IL-23 przez makrofagi CX3CR1+, aby zapobiec patogennej produkcji ILC3 IL-22 ( Bauché i in., 2018 ). Odwrotnie, anty-CD40 działa na DC CD103+, aby zwiększyć regulację CCR7 i migrować do węzła chłonnego krezkowego (mLN), gdzie ulegają apoptozie, zmniejszając liczbę RORγt+ Foxp3+ Tregs, które prowadzą do śmiertelnego zapalenia jelita grubego wywołanego przez komórki T ( Barthels i in., 2017 ). W tym badaniu przyjrzano się niezliczonym mechanizmom wrodzonej i adaptacyjnej odporności w jelitach, aby dokładnie określić, w jaki sposób THC zapobiega rakowi jelita grubego związanemu z zapaleniem jelita grubego.
THC osłabia postęp raka jelita grubego wywołanego zapaleniem okrężnicy, który jest związany ze zmniejszeniem produkcji IL-22 w mikrośrodowisku nabłonka
Aby wywołać raka jelita grubego, zastosowaliśmy dobrze ugruntowany model wstrzyknięcia substancji rakotwórczej, AOM (10 mg/kg, dootrzewnowo [ip]), po którym nastąpiły trzy cykle DSS. Podawanie THC (10 mg/kg, doustnie przez sondę) lub nośnika (VEH, PBS:EtOH:Tween-80, w stosunku 17:2:1) rozpoczęto równocześnie z pierwszym cyklem DSS i podawano dwa razy w tygodniu. Leczenie THC i VEH przerwano po trzecim cyklu DSS, aby zbadać wpływ THC na inicjację raka, a nie potencjalne bezpośrednie skutki THC na guzy. Postęp choroby i schemat eksperymentalny są szczegółowo opisane w ( Rysunek 1A ), ujawniając, że myszy, którym podano DSS + AOM w celu wywołania raka jelita grubego (grupa CC), ale leczono THC, straciły mniej na wadze w porównaniu z grupą CC + VEH ( Rysunek 1A ). Po zakończeniu badania grupa CC + VEH wykazywała guzy jelita grubego, podczas gdy CC + THC nie wykazywało guzów ( Rysunek 1 B). Wielkość śledziony była również zwiększona w grupie CC + VEH, podczas gdy leczenie THC doprowadziło do znacznego zmniejszenia wielkości śledziony ( Rysunki 1 C i 1D). Kolonoskopie i barwienie H&E jelit wykazały zmniejszenie nasilenia stanu zapalnego i indukcji guza w grupie CC + THC w porównaniu z CC + VEH ( Rysunki 1 E–1G). Ponieważ przewlekła regulacja w górę IL-22 prowadzi do rozwoju raka, a u myszy CC + THC nie rozwinęły się żadne guzy, przyjrzeliśmy się parametrom odpornościowym, w szczególności IL-22. IL-22 to cytokina produkowana głównie przez komórki Th22 i ILC w jelicie, która ma kluczowe znaczenie dla rozwoju, utrzymania i macierzystości raka jelita grubego wywołanego stanem zapalnym ( Sun i in., 2015 ; Kryczek i in., 2014 ; Kirchberger i in., 2013 ). Aby scharakteryzować populacje komórek odpornościowych, komórki blaszki właściwej okrężnicy (cLP) i komórki śródnabłonkowe (IEC) poddano analizie cytometrii przepływowej. Strategie bramkowania są szczegółowo opisane na rysunkach S1 A–S1D. Grupa CC + VEH miała znaczący wzrost liczby komórek Th22 CD4+IL-22+ w IEC okrężnicy w porównaniu z CC + THC, podczas gdy nie było zmiany w komórkach cLP CD4+ RORγt+ ( Rysunki 1 H–1J). Pobranie komórek z cLP i IEC i umieszczenie ich na noc w celu zebrania supernatantów wykazało, że wzrost IL-22 obserwowany w grupie CC + VEH w mikrośrodowisku okrężnicy pochodził konkretnie z komórek w IEC ( rysunki 1 K i 1L), a nie z cLP. Powyższe dane wykazały, że THC zapobiega rozwojowi raka jelita grubego w modelu DSS + AOM i że jest to związane ze spadkiem liczby IEC produkujących IL-22.
Aktywacja receptora kannabinoidowego hamuje postęp raka jelita grubego wywołanego zapaleniem okrężnicy poprzez zmniejszenie produkcji IL-22 w mikrośrodowisku nabłonka
Aby wywołać raka jelita grubego związanego z zapaleniem jelita grubego (CC), myszom podano pojedynczą dawkę AOM, ip (10 mg/kg), a następnie tydzień później rozpoczęto leczenie równocześnie z indukcją pierwszego cyklu zapalenia jelita grubego z 2% DSS w wodzie pitnej. Tygodniowe cykle DSS (2%) były kontynuowane przez 2 tygodnie regularnego picia wody przez 3 cykle trwające 9 tygodni. Leczenie VEH (10% EtOH w PBS-Tween-80), THC (10 mg/kg) lub kombinacją THC i CBD (10 mg/kg, oba) było podawane dwa razy w tygodniu aż do zakończenia ostatniego cyklu DSS, a następnie leczenie zostało przerwane w celu monitorowania wpływu kannabinoidów na indukcję raka, a nie bezpośredniego wpływu kannabinoidów na sam nowotwór. Myszy kontrolne były leczone dwa razy w tygodniu, ale choroba nie została wywołana (ctrl).
(A) Schemat przedstawiający procentową zmianę masy ciała, schemat leczenia i przebieg choroby. (n = 5, grupy kontrolne; n = 7–9 grup CC).
(B) Reprezentatywne zdjęcie i liczba guzów w każdym okrężnicy w momencie poświęcenia. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i są przedstawione jako średnia ± SEM; ∗∗p < 0,01, ∗∗∗p < 0,001 za pomocą testu Kruskala-Wallisa z testem wielokrotnych porównań Dunna.
(C i D) (C) Fotografia reprezentatywna i (D) ilościowe określenie masy śledziony u wskazanych myszy w momencie uśmiercenia.
(E i F) Kolonoskopie wykonywano przez cały okres trwania eksperymentu, a obrazy reprezentatywne przedstawiono w (E) i ilościowo w (F) (n = 5–8, grupy kontrolne; n = 8 grupy CC).
(G) Typowe obrazy H&E okrężnic w momencie uśmiercenia. cLP i frakcję komórek śródnabłonkowych (IEC) wyizolowano w momencie uśmiercenia i barwiono w celu wykrycia komórek CD4+ RORγt+ i CD4+ IL-22+.
(H) Typowe wykresy konturowe przedstawiające komórki Th17 CD4+ RORγt+ (bramka: żywe, CD45+ CD3+ CD4+) w cLP (dwa górne panele) i komórki Th22 CD4+ IL-22+ (bramka: żywe, CD45+ CD3+) we frakcji IEC (dwa dolne panele).
(I i J) Kwantyfikacja procentów z wykresów cytometrii przepływowej w (H), uzyskanych z (I) clP i (J) IEC (n = 3, grupy kontrolne; n = 3–8, grupy CC).
Komórki (K i L) 1 × 106 pochodzące z warstw (K) cLP lub (L) IEC ze wskazanych grup umieszczono na płytkach na noc, zebrano supernatanty i poddano testom ELISA na obecność IL-22 (n = 3–4).
Każdy symbol reprezentuje pojedynczą mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i są przedstawione jako średnia ± SEM NS, nieistotne: ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,001, ∗∗∗∗p < 0,0001 za pomocą dwukierunkowej analizy wariancji z testem wielokrotnych porównań Tukeya.
Komórki hematopoetyczne i niehematopoetyczne przyczyniają się do ochrony przed zapaleniem jelita grubego wywołanym przez THC, ale komórki hematopoetyczne są niezbędne do zmniejszenia produkcji IL-22
Aby ustalić, czy ekspresja CB na komórkach odpornościowych czy komórkach niehematopoetycznych była odpowiedzialna za korzystne efekty THC i określić mechanizm, poprzez który THC może zmniejszyć wydzielanie nowotworowe IEC IL-22, myszy dzikiego typu (WT) poddano mieloablacji i zrekonstruowano szpikiem kostnym (BM) myszy CB2KO (CB2KO→WT) lub WT BM (WT→WT), ponieważ zdecydowana większość ekspresji CB na komórkach odpornościowych to CB2. Następnie wywołaliśmy ostre zapalenie jelita grubego za pomocą wstrzyknięcia AOM i pojedynczego cyklu DSS, aby modelować wczesne stadia tumorigenezy przed makroskopowym rozwojem guza, ale gdzie występuje charakterystyczna utrata masy ciała, skrócenie jelita grubego i stan zapalny. W obu grupach, które otrzymywały THC, utrata masy ciała i skrócenie jelita grubego wywołane przez DSS zostały zniesione w porównaniu z kontrolami VEH, a skrócenie jelita grubego było bardziej wyraźne w grupie CB2KO → WT ( Rysunki 2 A i 2B). Co ciekawe, myszy CB2KO→WT, którym podano THC, miały zmniejszoną liczbę komórek Th22 w cLP w porównaniu z myszami VEH i WT→WT, ale tylko myszy WT→WT, którym podano THC, miały zmniejszoną liczbę komórek IEC Th22 ( rysunki 2 C i 2D). Wydzielanie IL-22 przez cLP ILC3 nie uległo zmianie pod koniec eksperymentu ( rysunek S1 E). Wpływ THC nie miał bezpośredniego wpływu na komórki Th22, ponieważ polaryzacja Th22 komórek naiwnych w obecności THC nie wpływała na wydzielanie IL-22 ( rysunek 2 E). Ponieważ wykazaliśmy, że THC może znieść indukcję aberracyjnej IL-22 w ostrych i przewlekłych modelach tumorigenezy i że wpływ ten jest niezależny od bezpośredniego wpływu THC na generację Th22, przeszliśmy do modelu, w którym stymulacja produkcji IL-22 jest pośredniczona przez produkcję prozapalnych cytokin przez komórki mieloidalne.
Komórki hematopoetyczne i niehematopoetyczne przyczyniają się do ochrony przed zapaleniem jelita grubego wywołanym przez THC, ale komórki hematopoetyczne są niezbędne do zmniejszenia produkcji IL-22
U myszy WT przeprowadzono ablację mieloblasty za pomocą dwóch dawek po 600 cGy w odstępie 3 godzin, a następnie przeprowadzono odbudowę układu odpornościowego poprzez przeniesienie komórek szpiku kostnego od myszy WT (WT → WT) lub myszy Cnr2−/−, z wyłączonym genem CB2 (CB2 → WT).
(A) Masa ciała w trakcie trwania choroby (n = 4–5).
(B) Długości okrężnicy na zakończenie eksperymentu.
(C i D) (C) Typowe wykresy konturowe przedstawiające komórki Th22 CD4+ IL-22+ (bramka: Żywe, CD45+ CD3+) we frakcji cLP IEC. (D) Ilościowe oznaczenie komórek ekspresujących CD4+ IL-22 w blaszce właściwej jelita grubego i frakcjach nabłonkowych (n = 4–5).
(E) Naiwne komórki T CD4+ wyizolowano od poszczególnych myszy i umieszczono w warunkach polaryzacji Th0 lub Th22 z VEH lub THC (10 μM) przez 3 dni. Zebrano supernatanty i poddano je testowi ELISA na obecność IL-22 (n = 6).
Każdy symbol reprezentuje pojedynczą mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i są przedstawione jako średnia ± SEM; NS, nieistotne statystycznie; ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,001, ∗∗∗∗p < 0,0001 metodą dwukierunkowej analizy wariancji z testem wielokrotnych porównań Tukeya.
Leczenie THC zmniejsza nasilenie zapalenia okrężnicy i całego układu wywołanego przez αCD40
Aby zbadać mechanizm, poprzez który THC zmniejsza produkcję IL-22 w jelitach, przeszliśmy do modelu zapalenia jelit przeciwko CD40 (αCD40). Wstrzyknięcie αCD40 powoduje ostre zapalenie jelit zależne od produkcji IL-22 ( Eken i in., 2014 ) i ogólnoustrojowy stan zapalny, który objawia się ostrą wyniszczającą chorobą i hiperaktywacją komórek zapalnych. Podawanie THC (10 mg/kg, doustnie przez sondę) rozpoczynające się 3 dni przed indukcją choroby nie mogło zmniejszyć utraty masy ciała związanej z początkiem choroby ( Rysunek S1 F). THC zmniejszyło splenomegalię występującą 7 dni po wstrzyknięciu αCD40 ( Rysunek 3 A) i zmniejszyło krążące prozapalne cytokiny IFNγ, IL-17A, IL-22 i TNFα w szczytowym momencie choroby 3 dnia; cytokiny związane z Th2 pozostały niezmienione ( Rysunki 3 B i 3C). Histologicznie THC zmniejszyło naciek komórek zapalnych i uszkodzenia spowodowane przez αCD40 w jelicie grubym i wątrobie ( rycina 3 D). Efekt ten był wyraźnie widoczny w jelicie grubym za pomocą kolonoskopii ( ryciny 3 E i 3F); uszkodzenie wątroby wywołane przez αCD40 mierzono, badając aktywność enzymatyczną aminotransferazy asparaginianowej (AST) i aminotransferazy alaninowej (ALT) w surowicy. Myszy αCD40, którym podano kontrole VEH, miały wyższą aktywność AST trzeciego dnia niż kontrole IgG, a myszy αCD40, którym podano THC, nie wykazywały tego wzrostu; jednak zarówno myszy leczone VEH, jak i THC miały wyższą aktywność ALT trzeciego dnia niż kontrole IgG ( ryciny 3 G i 3H).
Leczenie THC zmniejsza nasilenie zapalenia okrężnicy i całego układu wywołanego przez αCD40
Myszom podawano codziennie VEH lub THC (10 mg/kg) przez 3 dni przed dootrzewnowym wstrzyknięciem szczurzego przeciwciała anty-mysiego IgG (kontrola) lub szczurzego przeciwciała anty-mysiego αCD40 (200 μg, klon FGK4.5 w PBS), a leczenie kontynuowano przez 7 dni po wywołaniu choroby w celu monitorowania postępu ciężkości stanu zapalnego.
(A) Masa śledziony (n = 3–10).
(B) Trzeciego dnia pobrano krew poprzez krwawienie z oczodołu, oddzielono surowicę i poddano ją badaniu Legendplex w celu określenia poziomu cytokin pomocniczych T w surowicy (n = 3 w każdej grupie).
(C) Testy ELISA dla IFNγ, IL-22 i IL-17A z surowicy pobranej trzeciego dnia od myszy, którym podano leczenie lub VEH 3 dni przed wywołaniem choroby.
(D) Przykładowe obrazy H&E jelita grubego (górny rząd, powiększony 20X) i wątroby (dolny rząd, powiększony 4X).
(E i F) (E) Obrazy reprezentatywne z kolonoskopii wykonanych trzeciego dnia i ich ocena ilościowa (F) (n = 4 w każdej grupie).
(G i H) Ilościowe oznaczenie aktywności (G) AST i (H) ALT mierzonej w surowicy 3. i 7. dnia po wstrzyknięciu αCD40.
Każdy symbol reprezentuje pojedynczą mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla czterech niezależnych eksperymentów i są przedstawione jako średnia ± SEM. NS, nieistotne statystycznie; ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,005 w dwukierunkowej analizie wariancji z testem wielokrotnych porównań Tukeya.
Leczenie THC zmniejsza naciek komórek zapalnych jelita grubego i zwiększa liczbę Tregów Lamina Propria
Następnie staraliśmy się zbadać mechanizmy komórkowe w okrężnicy, poprzez które działa THC, aby zmniejszyć stan zapalny okrężnicy wywołany przez αCD40. Leczenie THC u myszy αCD40 skutkowało zmniejszeniem limfadenopatii krezkowej i zmniejszeniem odsetka komórek odpornościowych CD45+ migrujących do cLP, chociaż THC nie zmniejszyło całkowitej liczby komórek odpornościowych ( rysunki 4 A i 4B). Wstrzyknięcie αCD40 skutkowało ostrym uwolnieniem IL-22, które wbrew intuicji ochronnej roli IL-22 w modelach infekcji i chemicznych ( Gronke i in., 2019 ; Manta i in., 2013 ) jest patogenne na tych poziomach ( Eken i in., 2014 ). THC zmniejszyło wydzielanie IL-22 z komórek cLP u myszy αCD40 ( rysunek 4 C). Biorąc pod uwagę, że badanie aktywności cytokin z surowicy w dniu 3 ( Rysunek 3 B) nie wykazało wzrostu cytokin przeciwzapalnych IL-4, IL-10 lub IL-9 po leczeniu THC, rozważyliśmy mechanizmy supresji αCD40 niezależne od cytokin Th2. Zbadaliśmy komórki APC cLP i odkryliśmy, że THC zmniejszyło liczbę makrofagów cLP CD11b+ F4/80+, ale nie zmieniło liczby komórek DC cLP MHCII HI CD11c+ (strategie bramkowania opisano szczegółowo na rysunkach S1 A–S1D) ( Rysunki 4 D i 4E). Pomimo zmniejszenia liczby makrofagów cLP obserwowanego po leczeniu THC, markery aktywacji CD80 i CD86 nie uległy zmianie u myszy αCD40 otrzymujących THC, ale ekspresja komórek DC CD80 została zmniejszona po podaniu THC ( Rysunek S2 A–S2D). Aby ocenić, jaką rolę w chorobie odgrywają komórki DC cLP, scharakteryzowaliśmy podzbiory komórek DC, o których wiadomo, że silnie wpływają na równowagę zapalną jelit. Fenotypowaliśmy DC pod kątem ekspresji markerów powierzchniowych CD103 i CD11b, ponieważ wykazano, że DC z wyższą ekspresją CD103 odgrywają rolę w promowaniu odpowiedzi przeciwzapalnej poprzez indukcję Treg ( Coombes i in., 2007 ), podczas gdy DC z wyższą ekspresją CD11b są katalizatorami odpowiedzi zapalnych komórek T i ILC ( Persson i in., 2013 ). Leczenie THC spowodowało wzrost liczby CD103+ DC i równoczesny spadek CD103+ CD11b + DC w momencie uśmiercenia w porównaniu z myszami VEH ( Rysunek 4 F). Naturalne FoxP3+Helios + Treg wzrosły z αCD40 w porównaniu z IgG, a THC dodane do tego efektu w porównaniu z VEH ( Rysunek 4 G). W przypadku mLN leczenie THC spowodowało zmniejszenie liczby komórek CD8+ CTL wydzielających IFNγ i komórek CD4+ Th1 w porównaniu z VEH ( rysunki S2 E–S2H), natomiast nie zaobserwowano różnic w komórkach CD4+ wydzielających IL-17A, IL-10 lub IL-4 ani w fenotypie komórek DC ( rysunki S2 G–S2L).
Leczenie THC zmniejsza naciek komórek zapalnych jelita grubego i zwiększa liczbę Tregów Lamina Propria
(A) Bezwzględna liczba komórek węzłów chłonnych krezkowych (n = 3–6).
(B) Typowe histogramy nałożone na cytometrię przepływową, wartości procentowe i bezwzględna liczba komórek CD45+ z blaszki właściwej jelita grubego wskazanych myszy.
(C) Wyniki testu ELISA dla IL-22 z supernatantów cLP pobranych od wskazanych myszy w dniu 7 (n = 3–4).
(D–F) (D) Typowe wykresy konturowe cytometrii przepływowej makrofagów (bramka: żywe, CD45+) (n = 3–5), (E) komórek dendrytycznych (bramka: żywe, CD45+) (n = 5–8) i (F) podzbiorów komórek dendrytycznych cLP w 7. dniu (bramka: żywe, CD45+ MHCII HI CD11c+) (n = 3–9).
(G) Typowe wykresy konturowe cytometrii przepływowej dla n- i iTreg w cLP (bramka: Żywe, CD45+ CD3+ CD4+) (n = 3–8).
Każdy symbol reprezentuje pojedynczą mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla czterech niezależnych eksperymentów i są przedstawione jako średnia ± SEM. ns, nieistotne: ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,005 według dwukierunkowej analizy wariancji z testem wielokrotnych porównań Tukeya.
Aktywacja CB2 w komórkach dendrytycznych zmniejsza markery aktywacji i zwiększa produkcję TGF-β1
Zmianę fenotypu DC wywołaną przez THC obserwowano również w chemicznych modelach zapalenia okrężnicy wywołanego roztworem kwasu pikrylosulfonowego (TNBS) i DSS ( rysunki S3 A i S3B), zatem aby zobaczyć rolę DC w ochronie przed zapaleniem okrężnicy wywołanym przez THC, użyto naiwnych myszy. Myszom WT podano pojedynczą dawkę (1X) VEH lub THC, a 24 godziny później przeanalizowano cLP DC, ujawniając, że THC zwiększyło ekspresję DC CD103 i zmniejszyło ekspresję CD11b w warunkach podstawowych ( rysunek 5 A). Używając myszy WT, Cnr1 −/− i Cnr2 −/− , THC zwiększyło ekspresję CD103 u wszystkich genotypów myszy; jednak naiwne Cnr2 −/− miały obniżone ogólne poziomy cLP DC w porównaniu z myszami WT i Cnr1 −/− ( rysunek S4 A). Zgodnie z tym zauważyliśmy zależny od CB2 wzrost FoxP3+ Helios+ nTregs po podaniu THC ( Rysunek 5 B). Zwiększenie ekspresji receptora chemokiny 7 (CCR7) jest ważne dla transportu DC między tkankami limfatycznymi ( Schulz i in., 2009 ) i zostało zgłoszone jako jeden z mechanizmów, poprzez który DC mogą inicjować Treg w cLP. DC w mLN i cLP myszy, którym podano 1X THC, miały zmniejszoną ekspresję CCR7 w porównaniu z kontrolami VEH ( Rysunek 5 C). Innym mechanizmem, poprzez który CD103+ DC mogą wpływać na indukcję Treg, jest wydzielanie TGF-β1 ( Coombes i in., 2007 ), a supernatanty odzyskane z komórek cLP zebranych od myszy THC lub VEH 1X i wysianych na noc wykazały zwiększoną całkowitą produkcję TGF-β1 u myszy leczonych THC ( Rysunek 5 D). Nie zauważono wzrostu liczby komórek DC CD103+ w cLP po ostrym podaniu THC w mLN tych myszy ( Rysunek S4 B), ani nie zauważono wzrostu całkowitej produkcji TGF-β1 w mLN ( Rysunek S4 C). Aby wyizolować efekty THC konkretnie na komórki DC, wygenerowano komórki dendrytyczne szpiku kostnego (BMDC) przez dodanie GM-CSF i IL-4 do hodowli naiwnych komórek BM. VEH lub THC dodano in vitro w momencie polaryzacji, a za każdym razem zmieniano medium, aby ujawnić, że pod koniec hodowli komórki BMDC traktowane THC miały więcej całkowitego TGF-β1 w swoim supernatancie ( Rysunek 5 E). Chociaż w hodowlach traktowanych VEH i THC występowały równoważne procenty DC, hodowla BMDC jest mieszaną populacją komórek, więc 6. dnia hodowli BMDC oczyszczono DC CD11c+ i umieszczono je na noc bez dodatkowego VEH lub THC, odzyskano ich supernatant, a całkowite poziomy TGF-β1 zostały przeanalizowane i wzrosły w przypadku BMDC traktowanych THC w porównaniu z kontrolami VEH ( Rysunek 5 E). Zaobserwowaliśmy spadek ekspresji DC CD86, ale nie CD80 w hodowlach traktowanych THC; DC traktowane THC zmniejszyły proliferację komórek T CD4 i CD8 in vitro (Ryciny S4 D i S4E). Potwierdziliśmy te wyniki u myszy naiwnych poprzez sekwencjonowanie RNA pojedynczych komórek (scRNA-seq) komórek okrężnicy 24 godziny po podaniu VEH lub THC ( rycina S3 C). scRNA-seq pozwoliło na wyizolowanie komórek mieloidalnych z innych typów komórek, ujawniając, że THC zmniejsza ekspresję Cd80 i Cd86 w komórkach mieloidalnych , ale zwiększa ekspresję Cd40 ( rycina 5 F).
Aktywacja CB2 w komórkach dendrytycznych zmniejsza markery aktywacji i zwiększa produkcję TGF-β1
Naiwnym myszom podano VEH lub THC (10 mg/kg) jednorazowo, a po 24 godzinach pobrano cLP i mLN.
(A) Typowe wykresy konturowe cytometrii przepływowej podzbiorów komórek dendrytycznych w cLP.
(C) Typowe histogramy nałożone na cytometrię przepływową oraz mediana intensywności fluorescencji (MFI) ekspresji CCR7 w komórkach DC u wskazanych myszy w cLP lub mLN (n = 5).
(D i E) (D) Całkowite poziomy TGF-β1 z supernatantów komórek cLP pochodzących od myszy leczonych VEH lub THC przez 24 godziny (n = 5–6) i (E) BMDC leczone VEH lub THC po 7 dniach hodowli z leczeniem lub po 1 dniu hodowli po selekcji komórek CD11c+ (n = 3). Każdy symbol reprezentuje pojedynczą mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i przedstawione są jako średnia ± SEM. NS, nieistotne statystycznie: ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,005 metodą dwukierunkowej analizy wariancji z testem wielokrotnych porównań Tukeya.
(F) Wykres skrzypcowy scRNA-seq obrazujący ekspresję mRNA w klastrze komórek mieloidalnych z okrężnicy myszy leczonych VEH lub THC przez 24 godziny.
THC indukuje Tregs poprzez DC, aby zmniejszyć stan zapalny ogólnoustrojowy, ale nie jelitowy
Aby rozszyfrować, czy THC działało bezpośrednio na limfocyty T, czy poprzez DC, stymulując Treg, limfocyty T CD4+ wyizolowano od myszy naiwnych i umieszczono je w warunkach stymulacji limfocytów T (αCD3 i αCD28), polaryzacji Treg (αCD3, αCD28, IL-2 i TGF-β1) lub z komórkami APC CD11c+, z THC lub bez. Tylko wtedy, gdy limfocyty T były umieszczone na płytkach z komórkami APC CD11c+, THC zwiększyło odsetek Treg ( Rysunek 6 A); jednak THC działało bezpośrednio na spolaryzowane Treg, zwiększając ekspresję powierzchniową markerów funkcjonalnych TGF-β1 (LAP) i CD25 ( Rysunek 6 B), ale GARP, ICOS i CD223 (LAG-3) pozostały niezmienione ( Rysunek S4 F).
THC indukuje Tregs poprzez DC, aby zmniejszyć stan zapalny ogólnoustrojowy, ale nie jelitowy
Węzły chłonne i śledziony pobrano od myszy naiwnych, a komórki T CD4+ i APC CD11c+ wybrano poprzez oczyszczanie za pomocą kulek magnetycznych. Oczyszczone komórki T CD4+ umieszczono na płytkach na 5 dni w warunkach stymulacji komórek T: (αCD3 + αCD28), polaryzacji Treg: (αCD3 + αCD28 + IL-2 + TGF-β1) lub z oczyszczonymi komórkami CD11c+ w stosunku 5:1, CD4+:CD11c+, a następnie próbki od poszczególnych myszy potraktowano VEH lub THC (10 μM).
(A) Wykresy konturowe cytometrii przepływowej i ilościowe oznaczenie limfocytów Treg CD4+ FoxP3+ (bramka: żywe, CD4+) po 5 dniach hodowli w określonych warunkach (n = 5).
(B) Reprezentatywne nałożone histogramy wskazanych markerów z komórek CD4+ T leczonych THC lub VEH po polaryzacji Treg (bramka: Żywe, CD4+ FoxP3+) (n = 5). Każdy symbol reprezentuje sparowane porównanie z pojedynczej myszy, której komórki podzielono na leczone VEH lub THC. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i przedstawiono jako średnia ± SEM. NS, nieistotne: ∗p < 0,05, ∗∗p < 0,01 w teście t-Studenta. 10 dni przed indukcją choroby Treg zostały wyczerpane poprzez wstrzyknięcie dootrzewnowe szczurzego przeciwciała anty-mysiego CD25 (klon PC61, 250 μg/mysz) lub kontroli izotypowej. Myszy były wstępnie leczone VEH lub THC (10 mg/kg) przez 2 dni przed indukcją choroby poprzez wstrzyknięcie dootrzewnowe αCD40 lub kontroli IgG.
(C) ELISA na obecność cytokin w surowicy IFNγ, TNFα i IL-17A (n = 3–6).
(D) Masa śledziony na koniec eksperymentu (n = 3–6).
(E) Typowe wykresy konturowe cytometrii przepływowej i ilościowe oznaczenie wydzielających IL-22 komórek ILC3 (bramka: Żywa, linia-CD45 INT CD90.2 HI CD3- RORγt+) (n = 3–6).
(F) Wyniki testu ELISA dla IL-22 z supernatantów cLP pobranych od wskazanych myszy w dniu 7 (n = 3–5).
Każdy symbol reprezentuje pojedynczą mysz. Dane przedstawiono jako średnią ± SEM z jednego eksperymentu, który powtórzono 3 razy. NS, nieistotne: ∗p < 0,05, ∗∗p < 0,01 w dwukierunkowej analizie wariancji z testem wielokrotnych porównań Tukeya.
Aby ocenić rolę, jaką Treg odgrywają w osłabianiu procesów zapalnych αCD40 przez THC, Treg usunięto u myszy poprzez dootrzewnową iniekcję przeciwciała anty-CD25, które, jak wykazano, zmniejsza liczbę Treg 8 dni po pojedynczej iniekcji ( Setiady i in., 2010 ). Nie występowały one w mLN myszy leczonych przeciwciałami anty-CD25, ale nie u myszy kontrolnych IgG lub leczonych przeciwciałami αCD40+ IgG ( Rysunek S5 B). Leczenie THC lub VEH rozpoczęto 5 dni po podaniu przeciwciał anty-CD25 lub IgG i 2 dni przed αCD40. Ani myszy pozbawione Treg, którym podano αCD40, ani nie wykazywały większej utraty masy ciała w porównaniu z myszami αCD40+ IgG, ani THC nie zmieniało wyniszczenia ( Rysunek S5 A). THC zmniejszyło poziom IFNγ i TNFα w surowicy tylko u myszy αCD40+ IgG, nie u myszy αCD40 pozbawionych Treg; zmniejszenie poziomu Treg nie zmieniło zdolności THC do zmniejszania poziomu IL-17A w surowicy indukowanego przez αCD40 ( Rysunek 6 C). Utrata Treg zapobiegła zmniejszeniu masy śledziony obserwowanemu w przypadku THC+ αCD40 IgG ( Rysunek 6 D). Kolitogenna produkcja ILC3 IL-22 w modelu αCD40 jest częściowo regulowana przez Treg ( Bauché i in., 2018 ); jednak nasze wyniki wskazują, że THC zmniejsza wydzielające IL-22 ILC3, a także całkowitą produkcję IL-22 w okrężnicy niezależnie od Treg ( Rysunki 6 E i 6F).
THC zmniejsza markery aktywacji APC w jelicie grubym poprzez receptory kannabinoidowe
Ponieważ pokazaliśmy, że Tregs indukowane przez THC nie są pośrednikiem zapobiegającym zapaleniu okrężnicy, staraliśmy się przeanalizować inne zmiany komórek odpornościowych, które zachodzą po ostrym podaniu THC w warunkach naiwnych. Odkryliśmy, że THC nie wpływa na procent ILC: ILC2, NCR lub LTi ILC3 w cLP, ani nie zmienia się liczba makrofagów, chociaż występuje wzrost komórek tucznych FCεRI+ c-Kit+ w cLP po podaniu THC, a jest to pośredniczone przez CB2 ( Rysunki S5 C i S5D). Myszy leczone THC1X, których cLP wyizolowano i umieszczono na płytkach przez noc, wykazują jedynie niewielkie różnice w zakresie wzrostu produkcji IL-2 i IL-6 w porównaniu z myszami leczonymi VEH ( Rysunek S5 E). Biorąc pod uwagę globalną redukcję cytokin prozapalnych obserwowaną w przypadku THC po podaniu αCD40, przyjrzeliśmy się komórkom mieloidalnym w różnych tkankach, aby sprawdzić, w jaki sposób THC wpływa na ich immunogenność. cLP, mLN, śledziona, płuca, wątroba i mózg naiwnych myszy WT, Cnr1 −/− i Cnr2 −/− zostały pobrane, doprowadzone do zawiesiny pojedynczych komórek i umieszczone na noc w obecności VEH lub THC (10 μM), a następnie przeanalizowane cytometrią przepływową pod kątem interesujących markerów aktywacji APC: CD40, CD80, CD86, Ly-6C, Ly-6G, CD103, CX3CR1 i MHCII. Mapy cieplne przedstawiają medianę intensywności fluorescencji (MFI) dla każdego interesującego markera bramkowanego na makrofagach (Macs) lub DC z tej tkanki (strategie bramkowania dla poszczególnych tkanek przedstawiono na rysunkach S1 A–S1D) ( rysunek 7 A). Mapy cieplne są normalizowane dla każdego rzędu tak, aby zwiększone MFI silnie ekspresjonowanych markerów (np. MHCII na DC) nie przyćmiewało różnic w markerach o niższej ekspresji. Tylko markery, które wykazały istotne różnice w przypadku leczenia THC, są przedstawione graficznie. Stwierdziliśmy, że w tkance limfatycznej okrężnicy THC zmniejszyło ekspresję DC CD86 w mLN i cLP, ale zwiększyło ekspresję CD80 w cLP DC. Ekspresja CD40 wzrosła w obecności THC w mLN DC, ale spadła z THC w cLP DC, a CD103 wzrosła w cLP DC. W makrofagach jelitowych ekspresja CD86 i Ly-6C została zmniejszona w makrofagach mLN i cLP, podczas gdy w cLP ekspresja CD80 i CD103 wzrosła w makrofagach, jak widać w DC ( Rysunek 7 B).
THC zmniejsza markery aktywacji APC poprzez receptory kannabinoidowe
Myszy WT, Cnr1 −/− i Cnr2 −/−, dopasowane pod względem wieku i płci, poddano eutanazji, a błonę właściwą jelita grubego (cLP), węzły chłonne krezkowe (mLNs), śledziony (spl), wątroby, płuca i mózg sprowadzono do zawiesiny pojedynczych komórek, a następnie poddano działaniu ex vivo VEH lub THC (10 μM) przez 18 godzin przed analizą markerów aktywacji komórek prezentujących antygen (APC) metodą cytometrii przepływowej.
(A) Mapa cieplna medianowych natężeń fluorescencji (w kolejności malejącej) CD40, CX3CR1, CD80, CD86, Ly-6G, Ly-6C i MHCII na makrofagach i komórkach dendrytycznych z tkanek pochodzących od myszy WT, Cnr1 −/− lub Cnr2 −/− leczonych VEH lub THC. Natężenie mapy cieplnej zostało znormalizowane dla każdego rzędu/markera zainteresowania.
Każdy symbol reprezentuje inną mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i są przedstawione jako średnia ± SEM. ∗p < 0,05, ∗∗p < 0,01, ∗∗∗p < 0,005 metodą dwukierunkowej analizy wariancji z testem wielokrotnych porównań Tukeya.
W innych badanych tkankach zaobserwowano tendencję do zmniejszonej ekspresji wszystkich markerów u myszy Cnr2 −/− w porównaniu z myszami WT i Cnr1 −/− , co było najbardziej widoczne wśród komórek DC płuc ( rysunki S6 A–S6D). Co ciekawe, ani makrofagi, ani komórki DC w wątrobie nie wykazały zmiany markerów aktywacji z THC ( rysunek S6 A). Podobnie jak w jelitach, komórki DC płuc i śledziony wykazały spadek ekspresji CD86 pośredniczony przez CB2 w śledzionie ( rysunki S6 B i S6C). Ly-6C jest zmniejszone przez THC w komórkach DC płuc i mikrogleju, a CX3CR1 jest również zmniejszone w tych tkankach i w makrofagach śledziony ( rysunki S6 A–S6D). CD40 i Ly-6G są również zwiększone w mikrogleju z THC ( rysunek S6 D). Łącznie THC zmniejsza liczbę markerów aktywacji APC, zwłaszcza w barierach śluzówkowych jelit i płuc.
Aktywacja receptora kannabinoidowego 2 w komórkach mieloidalnych u myszy SCID pośredniczy w odporności jelita grubego na stan zapalny wywołany przez αCD40
W świetle danych dotyczących osłabiania zdolności stymulujących APC przez THC oraz faktu, że Tregs nie są mechanizmem, poprzez który THC zmniejsza produkcję IL-22 w okrężnicy, zainicjowaliśmy model αCD40 u myszy z niedoborem komórek T i B, aby zbadać, w jaki sposób modulacja komórek mieloidalnych przez THC wpływa na przebieg choroby. Podobnie jak u myszy WT BL/6, myszy SCID z niedoborem komórek T i B , którym podano αCD40, doświadczają ostrej utraty wagi, a leczenie VEH, THC lub THC plus antagonistami receptora CB AM251 (antagonista CB1) lub SR144528 (antagonista CB2) nie zmieniło wyniszczenia wywołanego przez αCD40 ( rycina S7 A). THC i THC + AM zmniejszyły stan zapalny okrężnicy, ale nie martwicze zapalenie wątroby ( ryciny 8 A–8C). Żadne leczenie nie zmniejszyło aktywności ALT w surowicy; jednak THC, THC + SR i THC + AM zmniejszyły aktywność AST w surowicy, co może sugerować zmniejszenie zaniku mięśni obserwowane w przypadku kannabinoidów ( rycina 8 D). Siedem dni po indukcji choroby, analizowano cLP DC i makrofagi, potwierdzając, że THC, THC + SR i THC + AM zwiększyły ekspresję CD103 DC, jak zaobserwowano u myszy WT ( ryciny 8 E i S7 B), ale na makrofagach tylko agonizm CB2 w grupie THC + AM zwiększył ekspresję CD103 przez makrofagi ( ryciny 8 E i S7 B). Komórki blaszki właściwej umieszczono na płytkach na noc i zebrano supernatanty, ujawniając, że leczenie THC i THC + AM zmniejszyło produkcję IL-23 i IL-1β, ale nie poziomy MCP-1, chociaż istniała tendencja do spadku ( rycina 8 F). U myszy naiwnych, którym podawano VEH lub THC przez 24 godziny, dane scRNA-seq wskazały, że podstawowa ekspresja Il1b i Ccl2 (MCP-1) nie uległa zmianie u myszy leczonych VEH i THC ( Rysunek S7 C). W tym modelu zapalenia okrężnicy wywołanego przez αCD40 z niedoborem komórek T, ILC3 są głównym źródłem okrężniczej IL-22, która nie uległa zmianie po jakimkolwiek leczeniu kannabinoidami ( Rysunek 8 G). Niemniej jednak, krążąca IL-22 została zmniejszona w grupach THC i THC + AM w porównaniu z grupami VEH i THC + SR w szczytowym momencie choroby i po zakończeniu badania ( Rysunek 8 H).
Aktywacja receptora kannabinoidowego 2 w komórkach mieloidalnych powoduje oporność jelita grubego na stan zapalny wywołany przez αCD40
Myszom SCID wstrzyknięto przeciwciała szczurze przeciwko myszom αCD40 (200 μg, klon FGK4.5 w PBS) i podawano im VEH, THC (10 mg/kg), THC+ SR144528 (THC+ SR, 10 mg/kg, oba) lub THC+ AM251 (THC + AM, 10 mg/kg, oba) codziennie przez 7 dni.
(A) Przykładowe obrazy H&E okrężnic (górny rząd, 4X) i wątroby (dolny rząd, 4X) u wskazanych myszy.
(B i C) (B) Ocena histologiczna obrazów H&E jelita grubego i (C) wątroby (n = 5).
(D) Aktywności ALT i AST mierzono w surowicy wskazanych myszy na zakończenie eksperymentu (n = 5).
(E) Mediana intensywności fluorescencji CD103 na komórkach DC i makrofagach z cLP myszy SCID , którym podano αCD40 i antagonistów VEH lub THC ± CB (n = 5).
(F) ELISA dla cytokin supernatantu cLP: IL-23, IL-1β i MCP-1 (n = 5).
(G) Wykresy konturowe cytometrii przepływowej komórek RORγt+ IL-22+ ILC3 w cLP wskazanych myszy po zakończeniu eksperymentu (n = 5).
(H) ELISA na obecność cytokiny IL-22 w surowicy w dniach 3 i 7 (n = 5).
Każdy symbol reprezentuje inną mysz. Dane pochodzą z jednego eksperymentu reprezentatywnego dla dwóch niezależnych eksperymentów i są przedstawione jako średnia ± SEM. ∗p < 0,05, ∗∗p < 0,01 w dwukierunkowej analizie wariancji z testem wielokrotnych porównań Tukeya.
Rozwój zapalenia jelita grubego jest wieloczynnikowy i powstaje w wyniku braku równowagi między barierą gospodarza, komórkami odpornościowymi i komensalami ( Lynch i Pedersen, 2016 ). Najnowsze dowody sugerują, że IBD rośnie globalnie ( Coward i in., 2019 ; Kaplan i Ng, 2016 ), a ponieważ mało prawdopodobne jest, aby genetyka była winowajcą w skali czasowej, musimy wziąć pod uwagę interakcje między środowiskiem, komensalami i reakcją naszych komórek odpornościowych na nie, jako czynnik wywołujący chorobę. Modulacja interakcji jelitowych między dowolnymi aktorami może przechylić szalę w kierunku promocji lub ochrony przed IBD. Wiele badań wykazało, że kannabinoidy wykazują potencjał w przechylaniu tej skali w kierunku tolerancji w celu ochrony przed IBD ( Singh i in., 2012 ; Storr i in., 2009 ; Ke i in., 2016 ; Kimball i in., 2006 ). Jednak dane dotyczące działania przeciwbakteryjnego kannabinoidów u ludzi nie odzwierciedlają tego, co zaobserwowano w modelach mysich ( Ambrose i Simmons, 2019 ; Naftali i in., 2013 ; Lahat i in., 2012 ). W modelu AOM + DSS raka jelita grubego leczenie THC zapobiegło rozwojowi raka i zmniejszyło liczbę pronowotworowych komórek Th22 wytwarzających IL-22, które infiltrowały śródnabłonkowo. IL-22 zyskała uwagę ze względu na swoje właściwości rakotwórcze u pacjentów ludzkich i w modelach zwierzęcych raka jelita grubego, promując macierzystość raka poprzez aktywację STAT-3 ( Sun i in., 2015 ; Kryczek i in., 2014 ; Kirchberger i in., 2013 ). Nasze wyniki wykazały, że THC osłabiało nowotworową IL-22 w mikrośrodowisku kolonocytów działając poprzez CB2 na komórki hematopoetyczne; jednakże efekt ten nie był wewnętrzny dla Th22, ponieważ THC nie wpływał bezpośrednio na izolowaną generację komórek Th22 in vitro . Stymulacja zarówno Th22, jak i ILC3 do produkcji IL-22 jest regulowana przez prozapalne cytokiny, takie jak IL-1β, IL-6, MCP-1 i IL-23 z lokalnych komórek APC ( Parks i in., 2016 ; Manta i in., 2013 ; Dudakov i in., 2015 ). W związku z tym zastosowaliśmy model zapalenia jelita grubego anty-CD40 u myszy dzikiego typu oraz u myszy z niedoborem komórek T i B, aby zbadać, w jaki sposób THC może oddziaływać na adaptacyjny i wrodzony układ odpornościowy, redukując poziom IL-22 w modelu, w którym jest on patogenny dla proksymalnego odcinka jelita grubego, niezależny od mikrobioty i nie polega na substancjach chemicznych egzogennych w pośredniczeniu w postępie choroby ( Uhlig i in., 2006 ).
Wstrzyknięcie przeciwciał anty-CD40 powoduje ostrą wyniszczającą chorobę i zapalenie okrężnicy zależne od wydzielania cytokin zapalnych TNFα, IL-12 i IL-23 ( Eken i in., 2014 ; Longman i in., 2014 ; Uhlig i in., 2006 ). Odkryliśmy, że w modelu zapalenia okrężnicy anty-CD40 THC zmniejsza stan zapalny układowy i okrężnicy poprzez redukcję makroskopowej patologii jelitowej i krążących prozapalnych cytokin IFNγ, TNFα, IL-17A i IL-22. Stan zapalny układowy wywołany przez przeciwciała anty-CD40 został zmniejszony przez THC, co zmierzono na podstawie zmniejszenia splenomegalii i biomarkerów uszkodzenia wątroby AST i ALT. Leczenie THC u myszy anty-CD40 spowodowało wzrost Treg w cLP, co było pośredniczone przez THC działające bezpośrednio na DC w celu zwiększenia ekspresji CD103 i zmniejszenia ekspresji CD11b, przekształcając je w bardziej tolerogenne komórki ze zwiększoną ekspresją TGF-β1 potwierdzoną przez scRNA-seq. Działając poprzez CB2, wykazaliśmy, że THC moduluje fenotyp cLP DC w kierunku bardziej przeciwzapalnego stanu, charakteryzującego się zmniejszoną ekspresją CD80 i CD11b u myszy z zapaleniem jelita grubego anty-CD40 in vivo . Nasze dane scRNA-seq komórek mieloidalnych cLP i analiza cytometrii przepływowej ex vivo naiwnych DC pochodzących ze szpiku kostnego i DC pochodzących z cLP, mLN, śledziony i płuc, wszystkie wykazały zmniejszenie ekspresji CD86 z THC. Interesujące odkrycie z naszej analizy cytometrii przepływowej globalnych markerów aktywacji APC ujawniło, że podczas gdy THC polaryzowało DC w kierunku bardziej tolerogennego fenotypu ze zwiększoną ekspresją CD103 i zmniejszoną ekspresją CD86, THC działało również na DC mLN i mikroglej, zwiększając ekspresję CD40. Zwiększone sygnały CD40 na DC CD103+ w celu regulacji CCR7, powodując ich przesunięcie do mLN i apoptozę ( Barthels i in., 2017 ). Biorąc pod uwagę, że widzieliśmy, że THC zmniejsza ekspresję CCR7 w cLP i mLN, jest mało prawdopodobne, aby powodowało to apoptozę DC; jednak CD40 działa z wcześniej aktywowanymi antygenem komórkami T CD40L+ w celu współstymulacji w dodatniej pętli sprzężenia zwrotnego, gdy napotkają znane antygeny ( Cong i in., 2000 ). Zwiększenie ekspresji CD40 przez THC w mLN, ale zmniejszenie ekspresji innych markerów aktywacji sugeruje, że THC może pobudzać odporność jelitową na zidentyfikowane antygeny, ale zmniejszać jego skuteczność w stymulowaniu odpowiedzi immunologicznej na nowe antygeny w cLP, co może wyjaśniać, dlaczego kannabinoidy są nieskuteczne w usuwaniu modeli infekcji zapalenia okrężnicy, ale są skuteczne w modelach chemicznych ( Eisenstein i Meissler, 2015 ; Newton i in., 2009 ; Karmaus i in., 2011 ). Zwiększenie ekspresji CD40 przez THC może być podobnie mechanizmem kompensacyjnym wyjaśniającym zmniejszenie ekspresji CD80 i CD86.
Komórki DC w cLP mogą promować ekspansję komórek T regulatorowych. Kilka badań zbadało ten mechanizm, sugerując, że komórki DC, głównie CD103+ DC, regulują CCR7, aby migrować do mLN, gdzie wydzielają TGF-β1 i kwas retinowy, aby indukować komórki T regulatorowe ( Schulz i in., 2009 ). Nasze wyniki wykazały, że podanie THC myszom naiwnym i po zapaleniu okrężnicy wywołanym przeciwciałami anty-CD40 spowodowało zmniejszenie ekspresji CCR7 na komórkach DC zarówno w cLP, jak i mLN, co sugeruje, że THC zmniejsza migrację komórek DC między tkanką limfatyczną przewodu pokarmowego. Ponadto odkryliśmy, że komórki BMDC leczone THC i komórki z cLP myszy leczonych THC wykazywały zwiększone poziomy TGF-β1. Łącznie te dane wskazują, że w jelitach THC powoduje, że DC zamiast migrować do mLN i indukować Treg, pozostają w cLP, obniżają poziom markera aktywacji CD86, wydzielają TGF-β1 i zwiększają odsetek Treg oraz wpływają na inne lokalne typy komórek poprzez działanie przeciwzapalne TGF-β1. TGF-β1 jest krytyczny dla homeostazy jelitowej. Globalne myszy TGF-β1 −/− rozwijają spontaniczne zapalenie jelita grubego w wieku około 3–4 tygodni, a swoiste dla DC wyłączenie TGF-β1 powoduje spontaniczne zapalenie jelita grubego ( Ihara i in., 2017 ; Ramalingam i in., 2012 ). DC są ważnym źródłem i aktywatorem TGF-β1 w jelitach, niezbędnym do kontrolowania różnicowania Treg i Th17 ( Ramalingam i in., 2012 ) oraz homeostazy kolonocytów ( Ihara i in., 2016 ). Wpływ THC na DC wynika z obserwacji, że leczenie THC zmniejsza aktywność APC u myszy ( Karmaus i in., 2011 ) oraz in vitro w komórkach ludzkich ( Roth i in., 2015 ). Aby zbadać wkład komórkowy pośredniczony przez THC w redukcję nasilenia przeciwciał anty-CD40, najpierw zubożyliśmy Treg poprzez wstrzyknięcie przeciwciał anty-CD25, ujawniając, że redukcja stanu zapalnego układowego wywołanego przez przeciwciała anty-CD40 przez THC jest częściowo pośredniczona przez Treg, ponieważ u myszy pozbawionych Treg, którym podano THC, krążące IFNγ i TNFα wzrosły z powrotem do poziomów kontrolnych, a splenomegalia nie została zmniejszona. Badanie przeprowadzone na myszach Rag2 −/− wykazało, że adopcyjne przeniesienie Treg nie zmieniło stanu zapalnego układowego ( Bauché i in., 2018).); jednakże miarą, której użyli do wyciągnięcia tego wniosku, była masa ciała, która również nie uległa zmianie w naszym badaniu z THC, z Tregami lub bez nich, chociaż krążące biomarkery aktywności choroby i masa śledziony uległy zmianie. Niemniej jednak, zapalenie okrężnicy wywołane przez przeciwciała anty-CD40 u myszy WT nie jest łagodzone przez Treg indukowane przez THC, ponieważ wyczerpanie Tregów nie osłabiło redukcji wydzielania IL-17A lub ILC3 IL-22 przez THC. Dane dotyczące cytokin wykazały również, że THC nie zwiększało innych cytokin przeciwzapalnych podzbioru Th: IL-4, IL-10 lub IL-9, co sugeruje, że zbawienne właściwości THC tkwiły w jego modulacji APC.
Dane z myszy SCID z niedoborem komórek T i B , którym wstrzyknięto przeciwciało anty-CD40, potwierdziły to, co obserwowano u myszy WT — komórki APC z jelita grubego są stymulowane przez przeciwciało anty-CD40, aby wywołać prozapalne IL-23 i IL-1β, które są redukowane przez agonizm CB2, oraz MCP-1, który wykazywał nieistotny statystycznie trend spadkowy przy leczeniu THC i THC + AM251. Analiza scRNA-seq komórek mieloidalnych WT cLP wykazała podobny spadek Il1b i Ccl2 po leczeniu THC. Zmniejszenie stanu zapalnego jelita grubego u myszy SCID αCD40 nie było odzwierciedlone przez zmniejszenie stanu zapalnego układowego ocenianego na podstawie patologii wątroby i poziomów ALT, co potwierdza to, co zaobserwowaliśmy w badaniach zubożających Treg u myszy WT. THC i kombinacja THC i obu antagonistów zmniejszyły poziom krążącej AST, co sugeruje, że THC może zmniejszyć zanik mięśni w tym modelu, albo poprzez nadmiarowy efekt CB, albo poprzez GPR55. Głównym odkryciem z tego eksperymentu było to, że THC działało na oba wartownicze APC w okrężnicy, DC i makrofagi, zwiększając CD103 i zmniejszając mediatory prozapalne, które stymulują produkcję IL-22. Chociaż nasze dane cLP nie wykazują różnic w wydzielaniu ILC3, poziomy IL-22 w surowicy są znacząco zmniejszone zarówno w dniu 3, jak i 7 przy leczeniu THC i THC + AM, co potwierdza rolę CB2 w zmniejszaniu wydzielania IL-22.
W tym badaniu wykazujemy, że agonizm THC wobec CB2 jest mechanizmem, poprzez który jelitowe APC przechodzą fenotypową zmianę w kierunku bardziej przeciwzapalnego fenotypu charakteryzującego się zwiększoną ekspresją CD103 i zmniejszoną CD86. Te APC są mniej przygotowane do wydzielania prozapalnych mediatorów IL-23, IL-1β i MCP-1, zamiast tego zwiększając TGF-β1 w celu indukowania Treg w lokalnym mikrośrodowisku. Treg są ważne dla systemowego rozwiązania stanu zapalnego wywołanego przez αCD40, zdolnego do zmniejszenia stanu zapalnego wątroby i splenomegalii; jednak w jelicie grubym, to poprzez działanie THC na DC i makrofagi, THC może zmniejszyć patogenne poziomy IL-22, aby chronić gospodarza przed nadmiernym stanem zapalnym i rakiem. Tutaj analizujemy, w jaki sposób aktywacja CB2 może udoskonalić fenotyp komórek mieloidalnych, aby znieść stan zapalny i zapobiec zapaleniu okrężnicy i rozwojowi raka okrężnicy.
Ograniczenia badania
Wykorzystując model nowotworu de novo i modele zapalenia jelita grubego wywołanego chemicznie i przeciwciałami, badanie to opisuje mechanizmy, poprzez które THC, działając poprzez CB2 na komórki APC w jelitach, ogranicza wydzielanie prozapalnych cytokin, które w przeciwnym razie stymulowałyby onkogenne uwalnianie Th22 i ILC3 IL-22. Określenie, czy redukcja IL-22 u myszy leczonych THC jest jedynym czynnikiem zapobiegającym rozwojowi CRC, będzie pouczające i warte oceny w przyszłych pracach. Chociaż badanie to oceniało działanie zapobiegające nowotworom THC, badania oceniające wpływ THC na rozwinięte nowotwory byłyby klinicznie istotne.
Układ endokannabinoidowy, w szczególności receptor kannabinoidowy 2 (CB2 u myszy i CNR2 u ludzi), ma kontrowersyjne implikacje patofizjologiczne w raku jelita grubego. Tutaj badamy rolę CB2 w potencjalizowaniu odpowiedzi immunologicznej w raku jelita grubego u myszy i określamy wpływ wariantów CNR2 u ludzi. Porównując myszy dzikiego typu (WT) z myszami z wyłączonym genem CB2 (CB2 -/- ), przeprowadziliśmy badanie spontanicznego raka u starzejących się myszy, a następnie wykorzystaliśmy model AOM/DSS raka jelita grubego związanego z zapaleniem jelita grubego i model dziedzicznego raka jelita grubego (Apc Min/+ ). Ponadto przeanalizowaliśmy dane genomiczne w dużej populacji ludzkiej, aby określić związek między wariantami CNR2 a zapadalnością na raka jelita grubego. Starzejące się myszy CB2 -/- wykazywały wyższą częstość występowania spontanicznych zmian przedrakowych w jelicie grubym w porównaniu z kontrolami WT. U myszy CB2 -/- i Apc Min/+ CB2 -/- leczonych AOM/DSS wystąpiła zaostrzona tumorigeneza i zwiększone populacje śledziony immunosupresyjnych komórek supresorowych pochodzących z mieloidów wraz ze zmniejszonymi komórkami T CD8+ przeciwnowotworowymi. Co ważne, potwierdzające dane genomiczne ujawniają istotny związek między niesynonimicznymi wariantami CNR2 a występowaniem raka jelita grubego u ludzi. Łącznie wyniki sugerują, że endogenna aktywacja CB2 tłumi tumorigenezę jelita grubego poprzez przesunięcie równowagi w kierunku komórek odpornościowych przeciwnowotworowych u myszy, a tym samym przedstawiają wartość prognostyczną wariantów CNR2 dla pacjentów z rakiem jelita grubego.
Według Narodowego Instytutu Zdrowia, rak jest jedną z głównych przyczyn zgonów na świecie, a liczba nowych przypadków raka rocznie ma wzrosnąć do 23,6 miliona do 2030 roku [ 1 ]. Konkretnie, rak jelita grubego jest jednym z najczęstszych nowotworów złośliwych obserwowanych w klinice i czwartą najczęstszą przyczyną zgonów związanych z guzem [ 2 ]. Przewlekły stan zapalny jest najważniejszym czynnikiem ryzyka ludzkiego raka jelita grubego (CRC). Pacjenci z chorobą zapalną jelit, chorobą Leśniowskiego-Crohna lub wrzodziejącym zapaleniem jelita grubego są narażeni na wysokie ryzyko i mają wyższy wskaźnik śmiertelności niż inni pacjenci z CRC [ 3 ].
Model myszy z rakiem jelita grubego związanym z zapaleniem okrężnicy (CAC) wywołany azoksymetanem/siarczanem sodu dekstranu (AOM/DSS) jest jednym z najczęściej stosowanych chemicznie indukowanych modeli CRC ze względu na jego powtarzalność i skuteczność [ 4 ]. Badania oparte na tym modelu potwierdziły znaczenie stanu zapalnego w rozwoju CRC i rzuciły światło na niektóre mechanizmy karcynogenezy jelita grubego związanej ze stanem zapalnym. W szczególności wyniki te wskazały na funkcję cytokin pro- i przeciwzapalnych w patologii CRC i udział środowiska immunosupresyjnego, a także na role określonych podgrup komórek układu odpornościowego [ 5 , 6 , 7 ].
Opracowano już szereg transgenicznych modeli myszy w celu odtworzenia czynników dziedzicznych, o których wiadomo, że są związane z rakiem jelita grubego u ludzi. Dobrze ugruntowany genetyczny model Apc Min/+ spontanicznie rozwija wiele nowotworów jelitowych i odzwierciedla ludzką rodzinną polipowatość gruczolakowatą i nowotwory jelita grubego. Te myszy niosą mutację w homologu myszy genu Apc , który jest kluczowym genem supresorowym nowotworu należącym do kanonicznej ścieżki sygnałowej Wnt. Model ten był wcześniej używany do ustalenia roli komórek supresorowych pochodzących z mieloidów (MDSC) w promowaniu nowotworu [ 8 , 9 , 10 ].
Komórki MDSC u pacjentów z rakiem jelita grubego korelują ze zmniejszonym przeżyciem i stały się główną przeszkodą w immunoterapii nowotworów [ 2 ]. Ta heterogeniczna populacja gromadzi się w układzie krążenia i narządach limfatycznych, a w szczególności w szpiku kostnym i miejscach występowania guzów u pacjentów z guzami. Wykazano, że te komórki MDSC promują inwazję, angiogenezę i unikanie odpowiedzi immunologicznej guza, co prowadzi do trwałego postępu nowotworu w wielu eksperymentalnych zwierzęcych modelach raka.
Endokannabinoidy i receptory endokannabinoidowe składają się na układ endokannabinoidowy (ECS) i współpracują, aby pośredniczyć w różnych procesach biologicznych i behawioralnych [ 11 ]. Receptory endokannabinoidowe, które są receptorami sprzężonymi z białkiem G (GPCR), są różnicowo ekspresjonowane w różnych tkankach, gdzie spełniają różnorodne funkcje [ 12 ]. ECS składa się z dwóch głównych endogennych ligandów, mianowicie N-arachidonoylethanolamine (AEA lub anandamid) i 2-arachidonoylglycerol (2-AG), oraz receptorów kannabinoidowych CB1 i CB2 (CNR1 i CNR2 u ludzi) [ 13 ]. CB1 jest ekspresowany głównie w neuronach, podczas gdy CB2 jest ekspresowany głównie w komórkach odpornościowych i kostnych [ 14 ]. Ze względu na stosunkowo wysoką ekspresję CB2 w komórkach odpornościowych, zasugerowano, że receptor ten pośredniczy w immunomodulacyjnych efektach endokannabinoidów [ 15 ]. Konkretna rola CB2 w raku nie została dokładnie zbadana w modelach z nienaruszonym układem odpornościowym, a kwestia, czy CB2 chroni przed rakiem czy go promuje, pozostaje kontrowersyjna [ 16 , 17 , 18 , 19 , 20 , 21 ]. Ta nierozwiązana kwestia, w połączeniu z naszymi wstępnymi danymi, które sugerują, że CB2 odgrywa ochronną rolę w tumorigenezie, dostarczyła motywacji do określenia roli endogennej aktywacji CB2 w stymulowaniu odpowiedzi immunologicznej gospodarza przeciwko rozwojowi guza. Tutaj przetestowaliśmy hipotezę, że CB2 odgrywa ochronną rolę w hamowaniu tumorigenezy, wykorzystując systemowy model knockoutu CB2 (CB2 −/− ) i dobrze ugruntowane modele raka jelita grubego u myszy immunokompetentnych i zbadaliśmy związek między wariantami genu CNR2 a rakiem jelita grubego w populacji ludzkiej.
2.1. CB2 −/− wiąże się ze zwiększonym ryzykiem wystąpienia samoistnego raka
Myszki dzikiego typu (WT) hodowane w warunkach SPF zazwyczaj nie rozwijają raka w pierwszym roku życia. Aby zbadać, czy CB2 przyczynia się do naturalnej odporności na spontanicznego raka, przeprowadziliśmy badanie histopatologiczne 14-miesięcznych samców i samic myszy WT i CB2 −/− . Dokładna analiza tkanek pobranych z jelita grubego i narządów rozrodczych (macicy, jajników i jąder) wykazała występowanie zmian nowotworowych i przednowotworowych u myszy CB2 −/− w sposób preferowany pod względem tkanki i płci. U samic myszy CB2 −/− zaobserwowano znaczący wzrost częstości występowania zmian nowotworowych i przednowotworowych (prawidłowych w porównaniu z nieprawidłowymi, dokładny test Fishera) i ich nasilenia (test U Manna-Whitneya) w jelicie grubym i macicy ( ryc. 1 A). Natomiast nie stwierdzono występowania zmian przedrakowych w jelicie grubym u samców myszy WT ani CB2 −/−, chociaż częstość występowania zmian przedrakowych w jądrach u myszy CB2 −/− była trzykrotnie wyższa (choć nieistotna statystycznie) w porównaniu do myszy WT tej samej płci ( Rysunek 1A ). Obserwacje te wskazują, że CB2 pełni rolę ochronną przez całe życie przed nowotworzeniem w przypadku różnych typów nowotworów, zwłaszcza u samic.
Kannabidiol (CBD) to nie odurzający i ogólnie dobrze tolerowany składnik konopi indyjskich, który wykazuje potencjalne korzystne właściwości w leczeniu szerokiego zakresu chorób, w tym zaburzeń sercowo-naczyniowych. Ze względu na złożony mechanizm działania CBD może oddziaływać na układ sercowo-naczyniowy na różne sposoby. W związku z tym dokonaliśmy przeglądu wpływu CBD na ten układ w zdrowiu i chorobie, aby określić potencjalne ryzyko sercowo-naczyniowych skutków ubocznych podczas stosowania CBD w celach medycznych i wellness oraz wyjaśnić jego potencjał terapeutyczny w chorobach sercowo-naczyniowych. Podawanie CBD zdrowym ochotnikom lub zwierzętom zwykle nie wpływa znacząco na parametry hemodynamiczne. Chociaż stwierdzono, że CBD wykazuje właściwości rozszerzające naczynia krwionośne i przeciwutleniające w leczeniu nadciśnienia, nie wpływa to na ciśnienie krwi u zwierząt z nadciśnieniem. Hipotensyjne działanie CBD zostało ujawnione głównie w warunkach stresowych. Wiele pozytywnych efektów CBD zaobserwowano w eksperymentalnych modelach chorób serca (zawał mięśnia sercowego, kardiomiopatia, zapalenie mięśnia sercowego), udaru mózgu, niedotlenieniowo-niedokrwiennej encefalopatii niedokrwiennej mózgu, zapalenia mózgu związanego z sepsą, powikłań sercowo-naczyniowych cukrzycy oraz uszkodzeń niedokrwiennych/reperfuzyjnych wątroby i nerek. W tych stanach patologicznych CBD zmniejsza między innymi uszkodzenia i dysfunkcje narządów, stres oksydacyjny i nitratywny, procesy zapalne i apoptozę. Niemniej jednak potrzebne są dalsze badania kliniczne, aby zalecić stosowanie CBD w leczeniu chorób sercowo-naczyniowych.
1. Wstęp
Cannabis sativa była używana od czasów starożytnych do celów rolniczych, ceremonialnych i leczniczych. W medycynie tradycyjnej roślina ta stosowana była jako środek przeciwbólowy, przeciwdrgawkowy, przeciwastmatyczny, przeciwmalaryczny czy przeciwreumatyczny. Konopie indyjskie zawierają ponad 700 różnych substancji chemicznych, wśród których wyróżnia się grupa związków zwana kannabinoidami. Kannabinoidy znajdujące się w konopiach indyjskich nazywane są fitokannabinoidami. Oprócz kannabinoidów pochodzenia roślinnego istnieją również kannabinoidy wytwarzane endogennie u ludzi lub zwierząt (tzw. endokannabinoidy) oraz kannabinoidy syntetyczne [ 1 , 2 ]. Zidentyfikowano ponad 100 fitokannabinoidów, a dwa najbardziej znane z nich (dla porównania zob.Tabela 1) to Δ 9 -tetrahydrokannabinol (THC, a dokładniej jego izomer (-)-trans) i kannabidiol (CBD). THC jest głównym psychoaktywnym składnikiem konopi indyjskich, a ze względu na swoje działanie odurzające marihuana, haszysz i olej haszyszowy są powszechnie używanymi nielegalnymi narkotykami. Natomiast CBD jest ogólnie uważane za substancję nieodurzającą (w literaturze często określane jest jako „niepsychoaktywne”, może jednak modulować objawy niektórych zaburzeń neuropsychiatrycznych, dlatego też określenie „nieodurzający” ma tendencję do być preferowane), nie powodując uzależnienia ani poważnych skutków ubocznych [ 1 , 3 , 4 , 5 ]. Ponadto może modulować działanie THC, a zatem zmniejszać lub nasilać (w zależności od dawki i stosunku CBD:THC) skutki uboczne THC [ 6 , 7 ].
CBD jest uważany za psychoaktywny ze względu na jego działanie przeciwlękowe, przeciwpsychotyczne i przeciwdepresyjne; 2 tetrad kannabinoidów charakteryzuje się hipolokomocją, hipotermią, katalepsją i antynocycepcją wywołaną przez THC i inne psychoaktywne kannabinoidy (agoniści CB 1 ) u myszy; 3 na podstawie badań przedklinicznych i klinicznych; zarejestrowane wskazania (USA i/lub UE) obejmują wyłącznie spastyczność w stwardnieniu rozsianym (Sativex ® ), padaczkę lekooporną – zespół Draveta i zespół Lennoxa-Gastauta (Epidiolex ® ), nudności i wymioty wywołane chemioterapią (Marinol ® , Syndros ® , Cesamet® , Canemes® ) i anoreksję związaną z AIDS (Marinol® , Syndros® ) ; 4 CBD jest częściowym agonistą GPR18 o niskiej skuteczności i antagonizuje działanie THC (CBD działa jako antagonista); ↑/↓ – zwiększenie/zmniejszenie; (+) – agonista lub dodatni modulator allosteryczny; (–) – antagonista, odwrotny agonista lub negatywny modulator allosteryczny; skróty: 2-AG: 2-arachidonoiloglicerol; 5-HT 1A, 2A, 3 : receptory serotoninowe typu 1A, 2A, 3; Abn-CBD: nieprawidłowy kannabidiol; AEA: anandamid; BP: ciśnienie krwi; CB 1, 2 : receptor kannabinoidowy typu 1, 2; D2 : receptor dopaminowy typu 2; EMT: transporter błonowy endokannabinoidów; FAAH: hydrolaza amidu kwasu tłuszczowego; FABP-3,-5,-7: białko wiążące kwasy tłuszczowe 3, 5, 7; GABA A : receptor kwasu γ-aminomasłowego typu A; GPR3, 6, 12, 18, 55: receptor 3, 6, 12, 18, 55 sprzężony z białkiem G; HR: tętno; PPAR-γ: receptor γ aktywowany przez proliferatory peroksysomów; TRPA1: członek 1 podrodziny ankyryn o potencjale przejściowym; TRPM8: przejściowy potencjał receptora, członek podrodziny melastatyny 8; TRPV1-4: przejściowe potencjalne receptory, członkowie podrodziny waniloidów 1-4; α1-, α1β-, α3-GlyR: receptor α1, α1β-, α3-glicyny; α1 – AR: α1 – receptor adrenergiczny; δ-, μ-OR: δ-, μ-receptor opioidowy.
Badania podstawowe i/lub kliniczne wykazały, że kannabidiol ma wielokierunkowe działanie (Tabela 1), takie jak między innymi działanie przeciwutleniające, przeciwzapalne, immunomodulujące, przeciwartretyczne, przeciwdrgawkowe, neuroprotekcyjne, prokognitywne, przeciwlękowe, przeciwpsychotyczne i antyproliferacyjne. Tym samym CBD posiada szeroki potencjał terapeutyczny, który obejmuje m.in. epilepsję, choroby neurodegeneracyjne (stwardnienie rozsiane, choroba Alzheimera, Parkinsona i Huntingtona), zaburzenia neuropsychiatryczne (depresja, zaburzenia lękowe, schizofrenia, zespół stresu pourazowego, zaburzenia ze spektrum autyzmu), zaburzenia żołądkowo-jelitowe. (nudności i wymioty, choroby zapalne jelit, zespół jelita drażliwego), choroby reumatyczne, choroba przeszczep przeciw gospodarzowi i nowotwory (omówione w innym miejscu: [ 5 , 6 , 17 , 30 , 31 , 32 , 33 ]). Jednakże większość tych wskazań wymaga dalszych badań w celu potwierdzenia skuteczności klinicznej.
Pierwszy lek wykorzystujący wyłącznie kannabidiol jako substancję czynną został zarejestrowany w czerwcu 2018 roku w USA pod nazwą Epidiolex ® (GW Pharmaceuticals, UK). Jest to preparat płynny zawierający CBD (100 mg/ml) pochodzenia roślinnego, wskazany do stosowania w leczeniu ciężkiej padaczki lekoopornej objawiającej się we wczesnym dzieciństwie, takiej jak zespół Draveta i zespół Lennoxa-Gastauta [ 28 ]. W Unii Europejskiej CBD ma obecnie status leku sierocego w przypadku kilku chorób, takich jak zespoły padaczkowe (wymienione powyżej i zespół Westa), asfiksja okołoporodowa, stwardnienie guzowate, choroba przeszczep przeciwko gospodarzowi i glejak (w połączeniu z THC w tym ostatnim przypadku) [ 34 ]. Z kolei w wielu krajach Europy dostępne są nabiximole (nazwa handlowa Sativex ® , GW Pharmaceuticals), czyli ekstrakt z konopi indyjskich zawierający CBD i THC w przybliżeniu w proporcji 1:1. Sativex ® podawany jest w postaci sprayu doustnego i został opracowany w celu łagodzenia objawów spastyczności u pacjentów ze stwardnieniem rozsianym [ 4 ]. Ponadto CBD i THC są obecne w marihuanie w różnych proporcjach, a do zastosowań medycznych opracowano pochodne (tzw. marihuana medyczna lub marihuana medyczna) [ 4 , 29 ]. Warto wspomnieć, że CBD występuje także w suplementach diety, kremach i balsamach do stosowania miejscowego oraz olejkach do waporyzacji [ 30,33 ] . Zwiększone zainteresowanie prozdrowotnymi i terapeutycznymi właściwościami tych produktów doprowadziło do ich powszechnego stosowania, co może wiązać się z potencjalnymi działaniami niepożądanymi lub interakcjami z współpodawanymi lekami.
Układ endokannabinoidowy, na który składają się endokannabinoidy, enzymy ich syntetyzujące i metabolizujące oraz receptory kannabinoidowe (CB 1 i CB 2 ) występuje w układzie sercowo-naczyniowym. Zarówno endogenne, jak i egzogenne kannabinoidy wywołują zmiany w układzie sercowo-naczyniowym ludzi i zwierząt [ 19 , 20 , 35 ]. Powikłania sercowo-naczyniowe, takie jak tachykardia i ostre incydenty wieńcowe, są powszechnie kojarzone z paleniem marihuany (skutki zależne głównie od THC) lub przyjmowaniem syntetycznych kannabimimetyków jako składnika dopalaczy [ 36 ]. Z kolei CBD pozbawione jest niekorzystnego wpływu na układ sercowo-naczyniowy. Ponadto sugeruje się, że ma potencjał terapeutyczny w leczeniu chorób układu krążenia, takich jak udar, zawał mięśnia sercowego, zapalenie mięśnia sercowego, kardiomiopatie i powikłania sercowo-naczyniowe cukrzycy, co wiąże się z właściwościami rozszerzającymi naczynia, kardioprotekcyjnymi, przeciwutleniającymi, przeciwzapalnymi i neuroprotekcyjnymi. CBD [ 23 , 24 ].
2. Biosynteza i farmakologia kannabidiolu
2.1. Struktura i biosynteza
Kannabidiol (patrz struktura chemiczna,Tabela 1), podobnie jak inne kannabinoidy, należy do grupy C21 (lub C22 w przypadku form karboksylowanych) terpenofenoli. W postaci kwasowej (patrz poniżej) kannabidiol jest głównym składnikiem włóknistych odmian konopi [ 1 , 4 ]. Po raz pierwszy został wyizolowany z konopi indyjskich przez Adamsa i wsp. w Wielkiej Brytanii [ 37 ] oraz z haszyszu przez Jacoba i Todda w USA [ 38 ] w 1940 r. Jednak jego strukturę chemiczną określili dopiero w 1963 r. izraelscy naukowcy Mechoulam i Schvo [ 39 ], a konfigurację absolutną cztery lata później Mechoulam i Gaoni [ 40 ].
Biosynteza i magazynowanie kanabidiolu i innych fitokannabinoidów zachodzi w włoskach gruczołowych występujących głównie na kwiatach żeńskich. Mniejsze ilości fitokannabinoidów wykrywa się także w liściach, łodygach, nasionach, korzeniach czy pyłkach. Oprócz rodzaju tkanki, stężenia związków bioaktywnych w konopiach indyjskich zależą od odmiany, warunków wzrostu, etapu wzrostu, czasu zbiorów i warunków przechowywania [ 3 , 41 ].
Biosynteza fitokannabinoidów (Rysunek 1) rozpoczyna się od syntezy dwóch związków prekursorowych – difosforanu geranylu (GPP) w szlaku 4-fosforanu 2-metyloerytrytolu (MEP) i kwasu oliwetolowego (OA) w szlaku poliketydowym. Część terpenowa kannabinoidów pochodzi z GPP, który powstaje w wyniku kondensacji difosforanu izopentenylu (IPP) i difosforanu dimetyloallilu (DMAPP) katalizowanej przez syntazę GPP [ 1 , 42 ]. IPP i DMAPP są izomerami (ulegają wzajemnej transformacji pod wpływem izomerazy IPP) i są syntetyzowane w plastydach szlakiem MEP. Prekursorami MEP są pirogronian i aldehyd 3-fosfogliceryny. Szlak cytozolowego kwasu mewalonowego (MEV) może być również źródłem IPP i DMAPP, ale w przypadku syntezy kannabinoidów jest to prawdopodobnie niewielkie [ 42 , 43 ]. Drugim ważnym prekursorem kannabinoidów jest OA odpowiedzialna za ich ugrupowanie fenolowe. Powstaje w wyniku kondensacji aldolowej heksanoilo-CoA z trzema cząsteczkami malonylo-CoA, a do tej przemiany potrzebne są dwa enzymy – syntaza oliwetolowa i cyklaza kwasu oliwetolowego, działające na powstały produkt pośredni, tetraketydowy. Heksanoilo-CoA jest produktem reakcji pomiędzy heksanianem (powstającym w wyniku biosyntezy i/lub degradacji kwasów tłuszczowych) i CoA, katalizowanej przez syntetazę heksanoilo-CoA. Z drugiej strony malonylo-CoA jest produktem karboksylacji acetylo-CoA przez karboksylazę acetylo-CoA [ 42 ].
Fuzja OA i GPP powoduje powstanie kwasu kannabigerolowego (CBGA) katalizowanego przez jego syntazę. CBGA jest uważane za główny związek prekursorowy fitokannabinoidów. Syntaza kwasu kannabidiolowego przekształca CBGA w kwas kannabidiolowy (CBDA). Podobnie kwas tetrahydrokannabinolowy (THCA) i kwas kannabichromenowy (CBCA) powstają w wyniku ich specyficznych syntaz. Kwasowe typy kannabinoidów są podatne na światło i ciepło i w wyniku nieenzymatycznej dekarboksylacji przekształcają się w formy obojętne – CBD, THC i kannabichromen (CBC) [ 1 , 42 ]. Należy zaznaczyć, że formy obojętne występują w rosnącej roślinie w niskich stężeniach, a dopiero podczas obróbki cieplnej surowca (spalanie, pieczenie) tworzą się w dużych ilościach [ 44 ].
2.2. Mechanizm akcji
Kannabinoidy wywierają swoje działanie poprzez interakcję z receptorami kannabinoidowymi CB 1 i CB 2 , odkrytą na początku lat 90-tych. Są to receptory metabotropowe związane z białkami G i/o , których pobudzenie powoduje hamowanie cyklazy adenylowej i stymulację kinaz białkowych aktywowanych mitogenami (MAPK) oraz (tylko dla CB 1 ) modulację kanałów wapniowych i potasowych. Co więcej, w działaniu receptorów CB1 mogą pośredniczyć białka Gq i Gs , niezależnie od białek G. Receptory CB 1 zlokalizowane są głównie w ośrodkowym układzie nerwowym, natomiast receptory CB 2 występują obficie w układzie odpornościowym. Zatem kannabinoidy mogą wywierać ważne prohomeostatyczne funkcje fizjologiczne poprzez modulowanie uwalniania neuroprzekaźników i odpowiedzi immunologicznych. Warto zauważyć, że wykazano obecność obu typów receptorów w całym organizmie, a ich ekspresja może zmieniać się w stanach patologicznych [ 17 , 22 , 27 ].
Kannabidiol ma niskie powinowactwo do receptorów kannabinoidowych (w stężeniach mikromolowych) [ 22 ]. Nie wywołuje efektów typowych dla stymulacji ośrodkowych receptorów CB 1 , takich jak hipoalgezja, hipotermia, katalepsja i obniżona aktywność motoryczna (tzw. tetrada kannabinoidowa), charakterystycznych dla THC [ 18 ]. Co więcej, CBD jest zdolne do antagonizowania działania agonistów receptora CB 1 /CB 2 (CP55940 i R-(+)-WIN55212) już przy stężeniach nanomolowych, a więc niższych niż wynikające z jego powinowactwa do tych receptorów [ 26 ]. Wykazano, że CBD jest odwrotnym agonistą receptora CB 2 [ 26 ] i negatywnym modulatorem allosterycznym receptorów CB 1 [ 12 ]. Pomimo braku agonistycznych właściwości receptorów CB 1 /CB 2 przez CBD, niektóre jego efekty są hamowane przez antagonistów/odwrotnych agonistów tych receptorów [ 8 , 45 ] lub nie są obecne u myszy z nokautem CB 1 [ 46 ]. Jest to najprawdopodobniej efekt pośredniego kannabinomimetycznego działania CBD, gdyż wykazano, że jego podawanie zwiększa stężenie endogennych kannabinoidów – anandamidu (AEA) [ 8 , 14 , 15 ] i 2-arachidonoiloglicerolu (2-AG) [ 45 ]. Mechanizmy tego efektu mogą obejmować zmniejszony rozkład i wewnątrzkomórkowy wychwyt endokannabinoidów. CBD hamuje główny enzym odpowiedzialny za rozkład AEA (oraz w mniejszym stopniu 2-AG) [ 27 ] – hydrolazę amidu kwasu tłuszczowego (FAAH) u gryzoni [ 9 , 16 ], ale nie u ludzi [ 10 ]. Co więcej, CBD hamuje wychwyt AEA, działając na domniemany transporter błonowy endokannabinoidów (EMT) [ 9 , 16 ] i/lub konkurując z AEA o wiązanie z białkami wiążącymi kwasy tłuszczowe (FABP-3, -5, -7), które stanowią wewnątrzkomórkowy system transportu endokannabinoidów [ 10 ].
Liczne badania wykazały, że CBD ma wiele efektów niezależnych od bezpośredniej lub pośredniej interakcji z receptorami CB 1 /CB 2 . Agonistyczne działanie CBD wykazano w odniesieniu do: przejściowego potencjalnego receptora, członka podrodziny ankiryn 1 (TRPA1) i członków podrodziny waniloidów 1–4 (TRPV1–4), receptora aktywowanego przez proliferatory peroksysomów γ (PPARγ), sierocego białka G receptor sprzężony – GPR18 (CBD jest częściowym agonistą, ale antagonizuje działanie THC) oraz receptory serotoninowe 5-HT 1A i 5-HT 2A (częściowy agonista). Ponadto CBD jest dodatnim allosterycznym modulatorem receptorów α1-, α1β- i α3-glicynowych (α1-, α1β- i α3-GlyR), μ- i δ-opioidowych (μ- i δ-OR) oraz γ-aminomasłowego. receptor kwasowy typu A (GABA A ). W przeciwieństwie do tego, CBD wykazuje działanie antagonistyczne w stosunku do receptora sierocego GPR55 (nawet postulowanego jako receptor CB 3 ), domniemanego receptora nieprawidłowego kannabidiolu (Abn-CBD; patrz poniżej) i potencjalnego receptora przejściowego, członka podrodziny melastatyny 8 (TRPM8). Ponadto jest negatywnym allosterycznym modulatorem receptora serotoninowego 5-HT 3 , receptora α 1 -adrenergicznego (α 1 -AR) i receptora dopaminy D 2 [ 11 , 17 , 21 ]. Ostatnio wykazano również, że CBD jest odwrotnym agonistą receptorów sierocych GPR3, GPR6 i GPR12 [ 13 ].
Oprócz bezpośredniej ekspozycji na szereg receptorów, CBD może oddziaływać także poprzez pośrednie zwiększanie stężenia związków biologicznie aktywnych. Oprócz wspomnianego już wpływu na poziom endokannabinoidów, CBD hamuje m.in. wychwyt adenozyny, tymidyny, glutaminianu, serotoniny, kwasu γ-aminomasłowego, dopaminy i noradrenaliny. Poziom serotoniny można także modulować poprzez hamowanie rozkładu jej prekursora, tryptofanu. Dodatkowo CBD wpływa także na metabolizm kwasu arachidonowego poprzez wpływ na fosfolipazę A2 ( PLA 2 ; stymulacja lub hamowanie w zależności od stężenia CBD), 5- i 15-lipooksygenazę (5-, 15-LOX; hamowanie) czy izoenzymy cyklooksygenazy (COX -1 i -2; hamowanie lub stymulacja). W konsekwencji wykazano zarówno zmniejszoną, jak i zwiększoną produkcję prostaglandyny E (PGE) [ 11 , 17 , 21 ]. Biorąc pod uwagę ścisłe powiązanie szlaków metabolicznych kwasu arachidonowego i endokannabinoidów (wspólne enzymy metaboliczne; kwas arachidonowy powstaje w wyniku rozkładu endokannabinoidów) [ 27 ], CBD może kompleksowo wpływać na powstawanie dużej grupy mediatorów – pochodnych kwasu arachidonowego i endokannabinoidów.
Podsumowując, CBD ma złożony profil farmakodynamiczny (Rysunek 2). Jednak w wielu przypadkach jego działanie występuje już przy bardzo wysokich stężeniach i dotychczas jedynie in vitro. Niemniej jednak tak złożony mechanizm działania może wyjaśniać szeroki potencjał terapeutyczny CBD.
Rysunek 2
Mechanizm działania kanabidiolu (CBD) [ 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 21 , 22, 26 , 27 , 45 , 46 ] . Podkreślono mechanizmy, poprzez które CBD oddziałuje na układ sercowo-naczyniowy [ 23 , 30 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 ] . 1 CBD jest częściowym agonistą GPR18 o niskiej skuteczności i antagonizuje działanie THC (CBD działa jako antagonista); skróty: 5-, 12-LOX: 5-,12-lipoksygenaza; 2-AG: 2-arachidonoiloglicerol; 5-HT 1A, 2A, 3 : Receptory serotoninowe typu 1A, 2A, 3; Abn-CBD: nieprawidłowy kannabidiol; AEA: Anandamid; A1 : Receptor adenozynowy typu 1; CB 1, 2 : Receptor kannabinoidowy typu 1, 2; COX-1,-2: Cyklooksygenaza 1, 2; D2 : Receptor dopaminy typu 2; EMT: transporter błonowy endokannabinoidów; EP 4 : Receptor prostaglandyny E 4; FAAH: hydrolaza amidu kwasu tłuszczowego; FABP-3,-5,-7: białko wiążące kwasy tłuszczowe 3, 5, 7; GABA A : receptor kwasu γ-aminomasłowego typu A; GPR3, 6, 12, 18, 55: receptor 3, 6, 12, 18, 55 sprzężony z białkiem G; IP: receptor prostacykliny; PGE: prostaglandyna E; PPAR-γ: receptor γ aktywowany przez proliferatory peroksysomów; TRPA1: Przejściowy potencjał receptora, członek podrodziny ankyryn 1; TRPM8: Przejściowy potencjał receptora, członek podrodziny melastatyny 8; TRPV1-4: Członkowie 1-4 podrodziny waniloidów o potencjale przejściowym; α1-, α1β-, α3-GlyR: receptor α1, α1β-, α3-glicyny; α1 – AR: α1 – receptor adrenergiczny; δ-, μ-OR: δ-, μ-Receptor opioidowy.
2.3. Farmakokinetyka
Istnieją różne drogi podawania kannabidiolu, z czego najczęstszą jest droga inhalacyjna (palenie, waporyzacja lub nebulizacja) i doustna (oleje, kapsułki, żywność i napoje wzbogacone w CBD). W zastosowaniach terapeutycznych można go także podawać w postaci sprayu na błonę śluzową jamy ustnej (Sativex ® ). Można go także podawać dożylnie, przezskórnie, doodbytniczo lub w postaci kropli do oczu [ 4 , 30 , 33 ]. Biodostępność jest zróżnicowana w zależności od drogi podania, np. dla drogi inhalacyjnej szacuje się ją na 31%, a maksymalne stężenie osiągane jest po 3–10 minutach od spożycia. Natomiast przy podaniu doustnym maksymalne stężenia osiągane są po 1–2 lub do sześciu godzinach od spożycia, a biodostępność wynosi poniżej 20%, ze względu na metabolizm pierwszego przejścia [ 4 , 58 ].
Niektórzy autorzy sugerują, że podanie doustne wiąże się z możliwą przemianą CBD w THC w kwaśnym środowisku żołądka (Rysunek 3). Taką konwersję stwierdzono w badaniach z symulowanym płynem żołądkowym [ 59 , 60 ]. Wydaje się jednak, że konwersja ta nie zachodzi in vivo u ludzi, o czym świadczy brak THC we krwi pacjentów, którzy przyjmowali doustnie nawet bardzo duże dawki CBD. Ponadto związek ten nie powoduje żadnych efektów psychologicznych, psychomotorycznych, poznawczych czy fizjologicznych typowych dla THC czy marihuany. Rozbieżność tę można wytłumaczyć faktem, że soki żołądkowe nie odzwierciedlają doskonale rzeczywistych warunków panujących w żołądku [ 61 , 62 ]. Badania na zwierzętach również wykazują sprzeczne dane. Hložek i in. [ 63 ] wykazali obecność THC we krwi szczurów po doustnym (a także podskórnym) podaniu CBD. Jednakże Palazzoli i in. [ 64 ] nie donieśli o braku THC we krwi szczurów, którym podano doustnie pojedynczą wysoką dawkę CBD po trzech i sześciu godzinach od podania. Podobnie nie stwierdzono THC u świnek morskich otrzymujących doustnie CBD przez pięć dni [ 65 ]. Zatem możliwa konwersja CBD na THC wydaje się wątpliwa.
Kannabidiol transportowany jest we krwi głównie w formie związanej z białkami, a około 10% CBD wiąże się z erytrocytami. Szybko ulega dystrybucji do wszystkich narządów dobrze ukrwionych, takich jak mózg, serce, płuca i wątroba. Objętość dystrybucji CBD wynosi około 32 l/kg. Ze względu na dużą lipofilowość może przy długotrwałym stosowaniu gromadzić się w tkance tłuszczowej [ 4 , 66 ].
Kannabidiol jest eliminowany poprzez metabolizm i wydalanie. CBD jest wydalane zarówno w stanie niezmienionym, jak i w postaci metabolitów z moczem i kałem [ 4 , 67 ]. Podawany okres półtrwania CBD u ludzi zależy od badania (różne dawki, drogi podawania) i może wahać się od około jednej godziny do pięciu dni [ 58 , 67 ]. Kannabidiol ulega biotransformacji składającej się z dwóch faz (Rysunek 3). Pierwsza zachodzi głównie w wątrobie, gdzie CBD ulega przemianom z udziałem izoenzymów cytochromu P450 (CYP). W badaniu z udziałem ludzkiego rekombinowanego CYP wykazano, że CYP1A1, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4 i CYP3A5 mogą metabolizować CBD, z czego CYP3A4 i CYP2C19 odgrywają dominującą rolę w mikrosomach wątroby [68 ] . Profile metaboliczne CBD różnią się w zależności od gatunku. U ludzi zidentyfikowano około 40 różnych metabolitów fazy I, a głównymi z nich są pochodne 7-karboksykannabidiolu (7-COOH-CBD). Aktywność farmakologiczna metabolitów fazy I jest różna, np. 7-hydroksy-kannabidiol (7-OH-CBD), podobnie jak CBD, hamuje wychwyt FAAH i AEA, podczas gdy 7-COOH-CBD nie wykazuje takiej aktywności; zarówno 7-OH-CBD, jak i 7-COOH-CBD nie są agonistami TRPV1. Zarówno CBD, jak i jego utlenione metabolity fazy I ulegają glukuronidacji, która jest główną reakcją fazy II [ 67 ].
Warto zaznaczyć, że CBD jest nie tylko substratem dla izoenzymów CYP, ale może również wpływać na ich aktywność. Wykazano, że CBD jest inhibitorem CYP1A1, CYP1A2, CYP1B1, CYP2C19, CYP2C9, CYP2D6, CYP3A4, CYP3A5 i CYP3A7 [ 11 ]. Z drugiej strony dłuższe przyjmowanie CBD może indukować ekspresję niektórych izoenzymów, co wykazano u myszy dla CYP3A i CYP2B10. CBD indukowało także ekspresję CYP1A1 w ludzkich komórkach wątroby. Zatem wpływ na aktywność/ekspresję izoenzymów CYP skutkuje możliwością interakcji CBD z innymi jednocześnie stosowanymi lekami [ 67 , 69 ]. W obszarze leków kardiologicznych opisano dotychczas jeden przypadek takiej interakcji. Jednoczesne stosowanie warfaryny (antykoagulantu) i CBD (Epidiolex ® ) nasilało działanie przeciwzakrzepowe (wzrost międzynarodowego współczynnika znormalizowanego, INR). Jak sugerują autorzy, mogło to wynikać z konkurencji o wiązanie i hamowanie przez CBD izoenzymów CYP zaangażowanych w metabolizm warfaryny [ 70 ]. Dlatego należy zachować ostrożność podczas jednoczesnego stosowania CBD z innymi lekami.
3. Wpływ kannabidiolu na układ sercowo-naczyniowy w warunkach fizjologicznych
Podawanie fito-, endo- i syntetycznych kannabinoidów ma różnorodny, czasem wielofazowy wpływ na ciśnienie krwi (BP) i częstość akcji serca (HR) w zależności od gatunku, drogi podania, obecności znieczulenia i innych warunków doświadczalnych [ 19 ]. Kannabinoidy mogą wpływać na czynność układu krążenia nie tylko poprzez receptory kannabinoidowe, ale szereg innych receptorów, zlokalizowanych zarówno w układzie nerwowym, jak i bezpośrednio w naczyniach krwionośnych i sercu [ 19 , 35 , 71 , 72 ]. Pobudzenie ośrodkowych receptorów CB 1 powoduje wzrost ciśnienia krwi, natomiast obwodowe receptory CB 1 zlokalizowane presynaptycznie na zakończeniach przed- i/lub zazwojowych neuronów współczulnych unerwiających serce i opór naczyniowy odpowiadają za hipotensyjne działanie kannabinoidów. Aktywacja receptorów CB 1 znajdujących się w mięśniu sercowym zmniejsza kurczliwość. Ponadto kannabinoidy mogą stymulować lub hamować odruch Bezolda-Jarischa, charakteryzujący się krótką i silną bradykardią oraz niedociśnieniem, za pośrednictwem receptorów TRPV1 i 5-HT3 zlokalizowanych na czuciowych włóknach nerwu błędnego [ 19 , 72 ]. Kannabinoidy w większości przypadków powodują rozszerzenie naczyń w izolowanych naczyniach krwionośnych lub perfundowanych łożyskach naczyniowych, chociaż obserwuje się również zwężenie naczyń. Efekty te wynikają z bezpośredniej aktywacji CB1 , TRPV1, PPAR i domniemanych śródbłonkowych receptorów kannabinoidowych. Ponadto w niektórych badaniach wykazano rolę receptorów CB 2 , GPR55 i 5-HT 1A w wazoaktywnym działaniu kannabinoidów [ 71 , 72 ]. Kannabinoidy mogą również aktywować sierocy GPR18 zlokalizowany obwodowo w naczyniach krwionośnych (GPR18 został przez niektórych autorów zdeorfanizowany jako śródbłonkowy receptor kannabinoidowy, patrz poniżej) i centralnie w rdzeniu brzuszno-bocznym przednim (RVLM), co powoduje rozluźnienie naczyń i niedociśnienie [ 35 , 73 ]. Kannabinoidy mogą również oddziaływać na układ sercowo-naczyniowy poprzez swoje metabolity, np. prostanoidy. Zatem rozluźnienie naczyń lub zwężenie naczyń wywołane przez kannabinoidy odbywało się pośrednio poprzez receptor prostaglandyny E 4 (EP 4 ) i receptor prostacykliny (IP) lub receptor prostaglandyny E 1 (EP 1 ) i receptor tromboksanu (TP), odpowiednio [ 19 , 71 ].
Poszukiwanie odpowiednich badań oceniających wpływ CBD na układ sercowo-naczyniowy w warunkach fizjologicznych przeprowadzono poprzez elektroniczne przeszukiwanie trzech baz danych (PubMed, Cochrane Library i EBSCO) od ich powstania do marca 2020 r. Słowa kluczowe wyszukiwania obejmowały: kannabidiol i układ sercowo-naczyniowy, hemodynamiczny, krew ciśnienie, tętno, przepływ krwi, naczynia krwionośne, serce, rozszerzenie naczyń lub działanie zwiotczające naczynia krwionośne. Ręcznie przeszukano także referencje z uwzględnionych badań.
Złożony mechanizm działania kannabidiolu pozwala na wielokierunkowe działanie na układ sercowo-naczyniowy. Jednakże dotychczasowe badania na zwierzętach i ludziach w dużej mierze wskazują na brak lub niewielki wpływ CBD podawanego doustnie (po), dożylnie (iv), dotętniczo, dootrzewnowo (ip), ośrodkowo lub poprzez inhalację (po ostrych i powtarzających się dawkowanie) na skurczowe (SBP), rozkurczowe (DBP) lub średnie (MBP) ciśnienie tętnicze krwi i/lub częstość akcji serca w warunkach fizjologicznych (Tabela 2) [ 7 , 15 , 49 , 55 , 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 , 92 , 93 , 94 , 95 , 96 , 97 ]. Potwierdzają to wyniki metaanalizy Sultana i wsp. [ 24 ], co wykazało brak wpływu CBD na HR i BP zarówno po podaniu ostrym, jak i przewlekłym (w tym drugim przypadku analizowano jedynie HR). Jednakże, jest kilka wyjątków. U ludzi CBD nieznacznie wzrosło (przy dawce 40 mg, ale nie 20 mg, podanej podjęzykowo) [ 98 ] lub obniżyło spoczynkowe ciśnienie krwi (600 mg, doustnie) po ostrym dawkowaniu [ 97 , 99 ], ale nie po wielokrotnym dawkowaniu (600 mg przez 7 dni, po) [ 97 ]. Odwrotnie, nie zaobserwowano tolerancji na hipotensyjne działanie CBD po jego przewlekłym doustnym dawkowaniu wzrastającym ze 100 do 600 mg/dzień przez 6 tygodni u pacjentów z dystonicznymi zaburzeniami ruchu [ 100 ]. Wykazano również, że CBD może zwiększać regionalny mózgowy przepływ krwi (CBF) [ 101 ]. Wpływ CBD na układ sercowo-naczyniowy u człowieka może zależeć nie tylko od dawki [ 98 ] i czasu podawania [ 97 ], ale także od sposobu dostarczania CBD. Zatem doustne CBD w dawce 90 mg nie miało wpływu na BP, HR i CBF, jednak ta sama dawka CBD kapsułkowana co TurboCBD TM (opatentowany preparat w kapsułkach zwiększających biodostępność CBD) zmniejszyła DBP i MBP oraz zwiększyła CBF [ 94 ]. U zwierząt CBD może również w różny sposób wpływać na parametry sercowo-naczyniowe. Zwiększał MBP i HR u psów znieczulonych pentobarbitalem [ 102 ], zmniejszał HR u przytomnych królików [ 103 ], zmniejszał MBP u myszy znieczulanych ketaminą i ksylazyną [ 104 ], nieznacznie podnosił SBP, DBP i HR u przytomnych szczurów [ 52 ] i zmniejszał MBP u szczurów znieczulonych pentobarbitalem [ 104 , 105 ]. U szczurów znieczulonych uretanem CBD podawane dożylnie nie miało wpływu na parametry hemodynamiczne [ 74] Jednakże po szybkim wstrzyknięciu może wywołać odruch Bezolda-Jarischa poprzez receptory TRPV1. Dodatkowo CBD osłabiło odruch Bezolda-Jarischa wywołany aktywacją receptora 5-HT 3 [ 52 ]. CBD może również modyfikować odpowiedź baroreceptorową po podaniu centralnym (do jądra łożyska prążka końcowego, BNST). Zatem wykazywał korzystny wpływ na odruchową reakcję bradykardyczną na wzrost ciśnienia krwi poprzez aktywację receptorów 5-HT 1A [ 47 ]. Po rdzeniu i wagotomii u szczurów znieczulonych uretanem, czyli po zniesieniu odruchów i wpływie ośrodkowego układu nerwowego na układ sercowo-naczyniowy, CBD spowodowało wzrost HR i ciśnienia skurczowego, natomiast spadek ciśnienia rozkurczowego prawdopodobnie wynikał z rozszerzenia naczyń [ 52 ]
Tabela 2
Wpływ kannabidiolu (CBD) in vivo na układ sercowo-naczyniowy w warunkach fizjologicznych 1 .
1 dotyczy wyłącznie układu sercowo-naczyniowego; 2 efekty zaobserwowane przy co najmniej jednej z badanych dawek; 3 pacjentów z jaskrą; 4 TurboCBD TM to opatentowany preparat w kapsułkach CBD zwiększający jego biodostępność (45 lub 90 mg CBD, 600 mg żeń-szenia amerykańskiego, 240 mg miłorzębu japońskiego , 150 mg organicznego oleju konopnego); 5 pacjentów z zaburzeniami obsesyjno-kompulsyjnymi; Szczur 6- rdzeniowy i z wagotomią; 7 pacjentów chorych na padaczkę; 8 pacjentów z dystonicznymi zaburzeniami ruchu; 9 pacjentów z chorobą Huntingtona; 10 pacjentów chorych na schizofrenię; 11 leczenie rozpoczynało się od dawki 200 mg/dobę i zwiększano ją stopniowo o 200 mg/dobę do dawki dobowej 800 mg/dobę (200 mg cztery razy na dobę) w ciągu pierwszego tygodnia (u niektórych pacjentów dawkę zmniejszono do 600 mg/dobę po dwa tygodnie ze względu na skutki uboczne); 11 szczurów kontrolnych z normotensją dla SHR (szczury Wistar-Kyoto); 12 szczurów kontrolnych z normotensją i szczurami z solą DOCA; ↑/↓/↔ — wzrost/spadek/brak zmian; skróty: 4-HHE: 4-Hydroksyheksenal; 4-HNE: 4-hydroksynonenal; 5-HT 1A,3 : Receptory serotoninowe typu 1A, 3; BNST: jądro łoża prążkowia końcowego; BP: ciśnienie krwi; CO: pojemność minutowa serca; DBP: rozkurczowe ciśnienie krwi; EKG: elektrokardiogram; EJT: czas wyrzutu lewej komory; FMD: dylatacja za pośrednictwem przepływu; HR: tętno; m.in.: dotętniczo; ic: Wewnątrzoponowo; ip: dootrzewnowo; iv: Dożylnie; MBP: Średnie ciśnienie krwi; MDA: dialdehyd malonowy; po: Per os, ustnie; PWV: prędkość fali pulsacyjnej; sl: podjęzykowo; SBF: przepływ krwi w skórze przedramienia; SBP: skurczowe ciśnienie krwi; SV: objętość skurczowa; THC –Δ 9 –Tetrahydrokannabinol; TPR: całkowity opór obwodowy; TRPV1: członek podrodziny waniloidów o potencjale przejściowym, receptor 4.
Wykazano działanie rozszerzające naczynia krwionośne CBD na izolowane naczynia ludzkie i zwierzęce, zarówno w warunkach fizjologicznych (Tabela 3) [ 48 , 56 , 106 ] oraz stany patologiczne (patrz niżej) i jest to prawdopodobnie najbardziej spójne działanie tego związku na układ sercowo-naczyniowy. Mechanizm działania CBD na naczynia jest złożony i zależy od badanego łożyska naczyniowego, nie obejmuje jednak receptorów kannabinoidowych [ 48 , 106 ], z wyjątkiem izolowanych ludzkich tętnic krezkowych, gdzie wykazano zależność od receptorów CB 1 [ 56 ] . Warto zauważyć, że CBD powoduje zależne od czasu rozszerzenie naczyń poprzez receptory jądrowe PPAR-γ [ 48 , 56 , 106 ]. Niemniej jednak działanie rozszerzające naczynia krwionośne CBD ex vivo w większości przypadków nie przekłada się na ogólnoustrojowe ciśnienie krwi (brak spadku ciśnienia krwi po podaniu CBD, patrz wyżej). Eksperymenty na szczurach z rdzeniem i wagotomią, u których CBD zmniejszyło DBP, wykazały, że działanie wazorelaksacyjne CBD in vivo może być maskowane przez napięcie neurogenne [ 52 ]. Badania na ludzkich komórkach śródbłonka aorty (Tabela 3) wykazało, że CBD zmniejszyło fosforylację N-końcowej kinazy c-Jun (JNK), czynnika jądrowego κB (NF-κB), rybosomalnej kinazy białkowej S6 (p70S6K) oraz przetwornika sygnału i aktywatora transkrypcji 5 (STAT5) oraz zwiększoną fosforylację Element odpowiedzi cAMP – białko wiążące (CREB), sygnał zewnątrzkomórkowy – kinaza regulowana 1/2 ( ERK1/2), kinaza białkowa B (Akt) i śródbłonkowa syntaza tlenku azotu (NOS). Te zmiany w fosforylacji białek wewnątrzkomórkowych mogą wyjaśniać właściwości CBD rozszerzające naczynia (ERK, Akt i śródbłonkowy NOS), antyangiogenne (p70S6K i STAT5) i przeciwzapalne (JNK i NF-κB) [56 ] . Kannabidiol wywiera także pozytywny wpływ na komórki mięśni gładkich naczyń (Tabela 3), których nieprawidłowa proliferacja i migracja są powiązane z rozwojem i postępem nowotworów i chorób układu krążenia. Zatem CBD hamowało proliferację i migrację komórek mięśni gładkich ludzkiej tętnicy pępowinowej oraz zwiększało aktywność enzymu cytoprotekcyjnego, oksygenazy hemowej-1. Temu ostatniemu efektowi towarzyszyła indukowana przez CBD produkcja reaktywnych form tlenu [ 107 ]. Jest to zaskakująca obserwacja, biorąc pod uwagę wiele badań wykazujących właściwości przeciwutleniające CBD [ 31 ]. Jednakże w innym badaniu CBD po podaniu chronicznie obniżyło markery stresu oksydacyjnego u szczurów z nadciśnieniem, ale wykazywało działanie prooksydacyjne u zwierząt kontrolnych z normotensją, szczególnie u szczurów Wistar-Kyoto (Tabela 2) [ 96 ].
Tabela 3
Wpływ kannabidiolu (CBD) in vitro i ex vivo na układ sercowo-naczyniowy w warunkach fizjologicznych 1 .
Gatunek
Narządy/komórki
Stężenie
Efekty 2
Bibliografia
Człowiek 3
Izolowane tętnice krezkowe (wstępnie zwężone U46619 4 i endoteliną-1)
0,1–100 μmol/l
-Rozszerzanie naczyń (efekt zależny od CB 1 , TRP, śródbłonka i NO; niezależny od CB 2 , receptora Abn-CBD, COX i kanałów potasowych)
-↓ fosforylacja JNK, NF-κB, p70S6K i STAT5;-↑ fosforylacja CREB, ERK1/2 (efekt zależny od CB 1 i TRPV 1 ), Akt (efekt zależny od CB 1 ) i śródbłonka NOS (efekt zależny od CB 1 )
1 dotyczy wyłącznie układu sercowo-naczyniowego; 2 efekty zaobserwowane przy co najmniej jednym z badanych stężeń; 3 pacjentów z nowotworem lub chorobą zapalną jelit; 4 agonista receptora tromboksanu; 5 pacjentów z rakiem płuc; agonista receptora 6α1 – adrenergicznego ; ↑/↓/↔ — wzrost/spadek/brak zmian; skróty: skróty: Abn-CBD: Abnormal-kannabidiol; AEA: Anandamid; Akt: Kinaza białkowa B; CB 1, 2 : Receptor kannabinoidowy typu 1, 2; COX: cyklooksygenaza; CREB: białko wiążące element odpowiedzi cAMP; EP 4 : Receptor prostaglandyny E 4; ERK1/2: kinaza regulowana sygnałem zewnątrzkomórkowym 1/2; GPR55: receptor 55 sprzężony z białkiem G; HO-1: oksygenaza hemowa-1; HR: tętno; IP: receptor prostacykliny; JNK: N-końcowa kinaza c-Jun; NF-KB: Czynnik jądrowy κB; BNO: Tlenek azotu i jego syntaza; PGE: prostaglandyna E; PPAR-γ: receptor γ aktywowany przez proliferatory peroksysomów; p70S6K: rybosomalna kinaza białkowa S6; ROS: reaktywne formy tlenu; SOD: dysmutaza ponadtlenkowa; STAT5: przetwornik sygnału i aktywator transkrypcji 5; TRP: potencjał receptora przejściowego; TRPV1: Przejściowy potencjał receptora, członek podrodziny waniloidów 1
Badania na izolowanych sercach, przedsionkach lub pojedynczych kardiomiocytach (Tabela 3) wskazują na bezpośrednie ujemne działanie inotropowe kanabidiolu [ 52 , 108 , 109 ]. W izolowanych sercach CBD może zmniejszać [ 108 ] lub nieznacznie zwiększać [ 110 ] częstość akcji serca, a także może mieć działanie proarytmiczne [ 108 ]. Badania na szczurach z rdzeniem i wagotomią wykazały, że CBD może pośrednio wpływać na serce poprzez wywieranie obwodowego efektu sympatykomimetycznego (objawiającego się wzrostem HR i SBP), prawdopodobnie ze względu na wpływ na uwalnianie i/lub wychwyt zwrotny noradrenaliny z układu współczulnego. zaciski [ 52 ]. W niektórych badaniach na ludziach i zwierzętach wykazano także zwiększenie częstości akcji serca (Tabela 2) [ 52 , 99 , 102 ]. Dodatkowo w metaanalizie Sultana i in. [ 24 ] podgrupa szczurów wykazała wzrost HR po podaniu CBD. Jednak w większości przypadków CBD nie wpływa na HR in vivo, co wskazuje (podobnie jak w przypadku naczyń krwionośnych), że efekty peryferyjne mogą być maskowane przez wpływy centralne.
Podsumowując, w warunkach fizjologicznych CBD ma minimalny wpływ na układ sercowo-naczyniowy. Dlatego nie niesie ze sobą zwiększonego ryzyka sercowo-naczyniowego, tak jak THC [ 36 ]. Ponadto CBD może osłabiać niektóre skutki wywołane THC w układzie sercowo-naczyniowym. U królików THC (3 mg/kg) i CBD (25 mg/kg) podane same (iv) obniżyły HR odpowiednio o około 40–50% i 10–20%. Jednakże wstępne leczenie CBD (25 mg/kg) zmniejszyło wielkość i czas trwania bradykardii wywołanej THC [ 103 ]. U ludzi odparowany THC (8 mg) prowadził do zatrucia i tachykardii. Niskie dawki CBD (4 mg) w połączeniu z THC wzmacniały jego działanie odurzające (ale nie wpływały na wzrost HR), podczas gdy wysokie dawki CBD (400 mg) łagodziły zarówno zatrucie wywołane THC, jak i tachykardię [ 7 ]. Zatem proporcje THC i CBD mogą być istotne dla ochronnego wpływu CBD na skutki sercowo-naczyniowe powodowane przez THC. W innym badaniu doustne CBD (200, 400 i 800 mg) nie zmienia subiektywnego, wzmacniającego i sercowo-naczyniowego działania palonej marihuany (1/2 papierosa zawiera ~800 mg konopi; 5,3–5,8% THC) [93 ] . Mieszanka CBD (1 mg/kg) i THC (0,5 mg/kg) (doustnie) nie zapobiegła tachykardii (w przeciwieństwie do lęku i innych efektów przypominających marihuanę) wywołanej przez samo THC podawane (0,5 mg/kg; doustnie) u ludzi [ 77 ] W badaniu klinicznym z THC (5 i 15 mg), niskimi (5,4 mg THC, 5,0 mg CBD) i wysokimi (16,2 mg THC, 15,0 mg CBD) dawkami Sativex ® nie stwierdzono znaczącej modulacji tachykardii i innych fizjologicznych objawów wywołanej przez CBD. zaobserwowano efekty wywołane przez THC [ 111 ]. Z drugiej strony CBD w równomolowych konsternacjach zmniejszyło wywołany THC wzrost HR oraz spadek ciśnienia tętna i przepływu krwi wieńcowej w izolowanych sercach szczurów [ 110 ].
4. Wpływ kannabidiolu na układ sercowo-naczyniowy w stanach patologicznych
Układ endokannabinoidowy nie wydaje się mieć istotnego znaczenia dla regulacji układu krążenia w warunkach fizjologicznych, gdyż zarówno inhibitory FAAH, jak i antagoniści receptora CB 1 nie wpływają znacząco na ciśnienie krwi u zwierząt z prawidłowym ciśnieniem [ 19 , 20 ]. Sytuacja ta zmienia się w stanach patologicznych, gdy często obserwuje się aktywację układu endokannabinoidowego [ 20 , 27 ]. Taka aktywacja może mieć działanie ochronne lub szkodliwe, np. wywołane endokannabinoidami rozluźnienie naczyń jest korzystne w przypadku nadciśnienia tętniczego, ale szkodliwe w przypadku wstrząsu septycznego lub nadciśnienia wrotnego [ 20 , 27 ]. Stany patologiczne mogą również modyfikować działanie podawanych kannabinoidów, np. reakcja depresyjna na THC jest silniejsza u pacjentów z nadciśnieniem niż u pacjentów z prawidłowym ciśnieniem [ 20 ]. Ponadto wpływ kannabinoidów na układ sercowo-naczyniowy można osiągnąć poprzez ich modulujący wpływ na procesy odpornościowe czy równowagę redoks zachodzącą za pośrednictwem receptorów kannabinoidowych i niekannabinoidowych. Kannabinoidy mogą powodować stres oksydacyjny i działanie prozapalne (głównie poprzez receptory CB 1 ), a także działanie antyoksydacyjne i przeciwzapalne (głównie poprzez receptory CB 2 ) [27.31]. Wiadomo, że kannabidiol ma właściwości przeciwutleniające (z pewnymi wyjątkami, patrz wyżej) i przeciwzapalne [ 6 , 30 , 31 , 32 , 33 ]. Zatem może mieć potencjał terapeutyczny w leczeniu różnych chorób układu krążenia, ponieważ stres oksydacyjny i stany zapalne są istotnymi elementami ich patogenezy.
4.1. Zmiany sercowo-naczyniowe wywołane stresem
Kannabidiol może działać jako środek przeciwlękowy w warunkach stresu zarówno w modelach zwierzęcych [ 55,82,87 ] , jak i u ludzi [ 80 ]. Stresujące sytuacje wiążą się ze wzrostem ciśnienia krwi i tętna, natomiast metaanaliza Sultana i wsp. [ 24 ] wykazało, że CBD eliminuje jedno i drugie. Zmniejszenie podwyższonych MBP i HR w szczurzych modelach stresu zaobserwowano po podaniu CBD dootrzewnowo [ 55 , 82 ], jak również centralnie do cisterna magna [ 87 ] lub BNST [ 90 ] (Tabela 4). Co ciekawe, wstrzyknięcie CBD do BNST spowodowało takie skutki jedynie w kontekście strachu uwarunkowanego kontekstowo u szczurów [ 90 ]. W ostrym stresie powstrzymującym CBD podawane do BNST (w tych samych dawkach) nie wpływało na MBP, a nawet nie zwiększało wzrostu HR wywołanego ograniczeniem [ 92 ]. Wykazano, że CBD wpływa na zmiany związane ze stresem w układzie sercowo-naczyniowym poprzez receptory 5-HT 1A [ 55 , 90 , 92 ]. Donoszono o sprzecznych wynikach u ludzi poddawanych różnym rodzajom stresu. Zatem CBD (300 lub 600 mg, doustnie) nie wpływało na ciśnienie krwi i/lub HR zwiększone w wyniku symulowanych wystąpień publicznych [ 80 , 112 ]. I odwrotnie, ostre podanie CBD (600 mg, doustnie) zmniejszyło lub wykazywało tendencję do obniżania ciśnienia krwi (i innych parametrów hemodynamicznych, patrzTabela 4) i zwiększone tętno podczas różnych stresujących warunków, takich jak mentalny test arytmetyczny, izometryczny test ściskania dłoni lub stres zimnem [ 99 ]. W innym badaniu ta sama dawka CBD zarówno po ostrym, jak i przewlekłym leczeniu (przez siedem dni) nieznacznie obniżyła SBP, ale nie miała wpływu na HR (i inne parametry sercowo-naczyniowe, patrzTabela 4) podczas ćwiczenia izometrycznego ściskania dłoni. Zatem tolerancja (obserwowana w warunkach spoczynku, patrz wyżej) na hipotensyjne działanie CBD podczas stresu nie rozwija się. Ponadto wykazano, że wielokrotne dawkowanie CBD zmniejsza sztywność tętnic i poprawia funkcję śródbłonka [ 97 ]. Podsumowując, CBD poza potencjalnym działaniem przeciwlękowym może wykazywać dodatkowo korzystne działanie hemodynamiczne w sytuacjach stresowych. Jednakże te efekty ochronne mogą przynajmniej częściowo wynikać z właściwości przeciwlękowych CBD. Warto zaznaczyć, że stres to prawdopodobnie stan, w którym wpływ CBD na parametry hemodynamiczne jest najbardziej wyraźny.
Tabela 4
Wpływ kannabidiolu (CBD) na zaburzenia sercowo-naczyniowe.
-↓ SBP (podczas testu warunków skrajnych), ↔ SBP (tuż przed i po teście warunków skrajnych)-↔ DBP, MBP, HR, CO, SV, EJT, TPR (tuż przed, w trakcie i po teście warunków skrajnych)
-↓ SBP (podczas testu warunków skrajnych), ↔ SBP (tuż przed i po teście warunków skrajnych)-↔ DBP, MBP, HR, CO, SV, EJT, TPR (tuż przed, w trakcie i po teście warunków skrajnych)-↓ sztywność tętnic (↓ PWV)-↑ funkcja śródbłonka (↑ FMD)
-↓ SBP, DBP, HR (odruch Bezolda-Jarischa indukowany przez receptory TRPV1; silniejszy niż w kontroli normotensyjnej)-↓ Odruch Bezolda-Jarischa wywołany aktywacją 5-HT3 ( ale nie TRPV1) (porównywalny z kontrolą normotensyjną)
0,1 mg/kg; 10 min przed okluzją i 10 min przed reperfuzją; IV
-↓ troponina I we krwi-↓ dysfunkcja lewej komory (zwiększenie SV, CO, EF, skurczowe pogrubienie ścian)-↑ przepływ krwi w obszarze z ubytkiem perfuzji-↓ wielkość zawału-↓ obrzęk mięśnia sercowego i niedrożność mikronaczyń-↓ naciek neutrofilów w sercu-↓ apoptoza w sercu
-↓ troponina T i CK-MB w surowicy-↓ zmiany histopatologiczne w sercu-↓ stres oksydacyjny i nitratywny w sercu-↓ zapalenie serca-↓ apoptoza w sercu-↓ Ekspresja NF-κB w sercu-↓ Ca i ↑ Zn i Se w sercu
-↓ CK i LDH w surowicy-↓ dysfunkcja serca-↓ stres oksydacyjny i nitratywny w sercu-↓ upośledzona funkcja mitochondriów serca i biogeneza-↓ aktywacja MMP2 i MMP9 w sercu-↓ śmierć komórek w sercu-↓ zapalenie serca
-↓ dysfunkcja serca (poprawa funkcji skurczowej i cofnięcie dysfunkcji rozkurczowej oraz sztywność mięśnia sercowego)-↓ zwłóknienie mięśnia sercowego-↓ stres oksydacyjny i nitratywny w sercu-↓ zapalenie serca-↓ naciek komórek jednojądrzastych w sercu-↓ martwica serca
4. Udar mózgu, noworodkowa encefalopatia niedotlenieniowo-niedokrwienna , zapalenie mózgu związane z sepsą
Prosiaczek (noworodek)
Niedotlenienie i niedrożność tętnic szyjnych (20 min) + okres po HI (6 godz.); model noworodkowego HIE
0,1 mg/kg; 15 minut i 240 minut po HI; IV
-↓ mózgowe upośledzenie hemodynamiczne i metaboliczne-↑ aktywność mózgu (amplituda EEG)-↓ drgawki-↓ utrata neuronów i degeneracja neuronów w korze i hipokampie-↓ troponina T we krwi-↓ Wywołany przez HI spadek MBP i wzrost HR-↓ Gazometria wywołana przez HI i zaburzenia oddechowe
Niedotlenienie i niedrożność tętnic szyjnych (40 min) + okres po HI (6 godz.); model noworodkowego HIE
1 mg/kg; 30 minut po HI; IV
-↓ upośledzenie aktywności mózgu-↓ martwica neuronów w korze mózgowej-↑ liczba astrocytów w korze mózgowej-↓ ekscytotoksyczność w korze mózgowej-↓ stres oksydacyjny w korze mózgowej-↓ zapalenie układu nerwowego w korze mózgowej-↓ Spadek MBP wywołany HI-↔ pCO2 we krwi i obniżone pH krwi-↔ CO (efekty zależą od CB 2 i 5-HT 1A )
Niedotlenienie i niedrożność tętnic szyjnych (20 min) + okres po HI (6 lub 72 godz.); model noworodkowego HIE
0,1 mg/kg; 15 minut i 240 minut po HI; IV
-↑ aktywność mózgu (amplituda EEG)-↓ upośledzenie metabolizmu mózgu-↓ upośledzenie funkcji neurobehawioralnych-↓ zmiany histopatologiczne w mózgu-↓ Komórki TNF-α-dodatnie w mózgu-↓ S100B (marker uszkodzenia astrocytów) i enolaza specyficzna neuronalnie w płynie mózgowo-rdzeniowym
Niedotlenienie i niedrożność tętnic szyjnych (40 min) + okres po HI (6 godz.); model noworodkowego HIE
1 mg/kg; 30 minut po HI; IV
-↓ upośledzenie aktywności mózgu-↓ martwica neuronów w korze mózgowej-↓ upośledzenie metabolizmu neuronalnego w korze mózgowej-↓ apoptoza w korze mózgowej-↓ ekscytotoksyczność w korze mózgowej-↓ stres oksydacyjny w korze mózgowej-↓ zapalenie układu nerwowego w korze mózgowej-↓ Spadek MBP wywołany HI-↔ pCO2 we krwi i obniżone pH krwi-↔ CO-↑ korzystny wpływ hipotermii na toksyczność wywołaną HI, zapalenie układu nerwowego, stres oksydacyjny i uszkodzenie neuronów w korze mózgowej
Niedotlenienie + okres po niedotlenieniu (9,5 godz.); model noworodkowego HIE
1 mg/kg; 30 min po niedotlenieniu; IV
-↔ zmiany neuropatologiczne w korze mózgowej, hipokampie, istocie białej i móżdżku-↔ markery stresu oksydacyjnego w moczu-↔ zapalenie układu nerwowego w korze mózgowej i hipokampie-↔ apoptoza w korze mózgowej-↔ ekscytotoksyczność w hipokampie-↔ upośledzenie metabolizmu neuronalnego w korze mózgowej-↔ hemoglobina, mleczany, glukoza i troponina T we krwi-↔ NGAL w moczu (↓ NGAL w moczu, gdy zastosowano hipotermię)-↔ S100B (marker uszkodzenia astrocytów) w płynie mózgowo-rdzeniowym-↔ Indukowany HI spadek MBP i wzrost HR-↔ Nieprawidłowości w gazometrii wywołane przez HI-↔ korzystne działanie hipotermii
Niedotlenienie + okres po niedotlenieniu (6 godz.); model noworodkowego HIE
1 mg/kg; 30 min po niedotlenieniu; IV
-↓ Spadek MBP wywołany HI-↑ aktywność mózgu (amplituda EEG)-↓ śmierć neuronów w mózgu ↓ zapalenie nerwów w mózgu-↓ stres oksydacyjny w mózgu-↓ obrzęk płuc i zmiany histologiczne oraz stany zapalne w płucach (wszystkie powyższe objawy zależą od 5-HT 1A )-↓ wymiana gazowa w płucach i ↑ całkowita pojemność płuc (działanie jest niezależne od 5-HT 1A )-↔ stres oksydacyjny w płucach-↔ CO i gazy krwi
Niedotlenienie + okres po niedotlenieniu (9,5 godz.); model noworodkowego HIE
50 mg/ kg5 ; 30 min po niedotlenieniu; iv ponad 15 min.
-↓ MBP (istotne)-↔ HR, temperatura ciała, hemoglobina, mleczan i troponina T we krwi-↔ S100B (marker uszkodzenia astrocytów) w płynie mózgowo-rdzeniowym-↔ zmiany neuropatologiczne w mózgu-↔ ekscytotoksyczność w mózgu-↔ upośledzenie metabolizmu neuronalnego w mózgu
okluzja MCA (1 godz.) + reperfuzja (1 dzień); model udaru
50, 100 lub 200 ng; przez 5 dni przed okluzją; icv
-↓ całkowita objętość zawału półkuli mózgu, kory mózgowej, kory gruszkowatej, ciała migdałowatego i prążkowia-↓ Receptor TNF 1 i NF-KB łącznie w półkuli mózgu, korze i prążkowiu
okluzja MCA (4 godz.) + reperfuzja (20 godz.); model udaru
0,1; 1; 3 lub 10 mg/kg; bezpośrednio przed okluzją i 3 godziny po rozpoczęciu okluzji; ip
-↓ objętość zawału (efekt zależny od 5-HT 1A ; niezależny od CB 1 i TRPV1)-↑ CBF podczas okluzji (efekt zależny od 5-HT 1A )-↔ MBP, HR (2 godziny po rozpoczęciu okluzji)-↔ gazometria i hematokryt przed reperfuzją
okluzja MCA (4 godz.) + reperfuzja (20 godz. lub 3 dni); model udaru
3 mg/kg; bezpośrednio przed okluzją i 3 godziny po rozpoczęciu okluzji; ip
-↓ objętość zawału (20 godzin lub 3 dni po okluzji)-↔ gazometria, hematokryt, K i Na we krwi (przed reperfuzją)-↔ MBP, HR (przed reperfuzją)-↔ temperatura w odbycie (przed reperfuzją)-↑ CBF podczas okluzji i przez 1 godzinę po okluzji-↓ Aktywność MPO w mózgu (1 i 20 h po okluzji; działanie jest niezależne od CB 1 i CB 2 )-↓ Komórki MPO-dodatnie w prążkowiu (20 godzin lub 3 dni po okluzji)-↑ koordynacja ruchowa (3 dni po okluzji)
okluzja MCA (4 godz.) + reperfuzja (20 godz.); model udaru
0,1; 1 lub 3 mg/kg; bezpośrednio przed okluzją i 3 godziny po rozpoczęciu okluzji; ip
-↓ objętość zawału (efekt jest niezależny od CB 1 i CB 2 )
3 mg/kg; bezpośrednio przed okluzją lub 3, 4, 5, 6 h po rozpoczęciu okluzji; ip
-↓ objętość zawału-↔ ekscytotoksyczność w korze w ciągu 2 h od wystąpienia okluzji (CBD podane bezpośrednio przed okluzją)-↓ Aktywność MPO w mózgu (CBD podane 6 h po wystąpieniu okluzji)
Mysz
okluzja MCA (4 godz.) + reperfuzja (20 godz.); model udaru
0,1; 1 lub 3 mg/kg; bezpośrednio przed okluzją i 3 godziny po rozpoczęciu okluzji; ip
-↓ objętość zawału (efekt niezależny od CB 1 i CB 2 ; zależny od 5-HT 1A )-↑ CBF podczas okluzji (efekt zależny od 5-HT 1A )-↔ gazometria, hematokryt, K i Na we krwi przed reperfuzją-↔ Ekspresja CB 1 w korze, prążkowiu i podwzgórzu
3 mg/kg; przez 14 dni przed okluzją + bezpośrednio przed okluzją i 3 godziny po wystąpieniu okluzji; ip
-↓ objętość zawału (w zależności od 5-HT 1A )-↑ CBF podczas okluzji (efekt zależny od 5-HT 1A ) (efekty są porównywalne z obserwowanymi w grupie nieleczonej CBD przez 14 dni) -↔ temperatura w odbycie po 1 godzinie od wystąpienia okluzji-↔ gazometria, hematokryt, K i Na we krwi przed reperfuzją-↔ Ekspresja CB 1 w korze, prążkowiu i podwzgórzu
Mysz
okluzja MCA (4 godz.) + reperfuzja (20 godz.); model udaru
0,1; 1 lub 3 mg/kg; bezpośrednio przed okluzją i 3 godziny po rozpoczęciu okluzji; ip
-↔ MBP, HR, pH, pCO 2 , hematokryt, Na, K, poziom glukozy we krwi, temperatura ciała (przed reperfuzją)-↓ objętość zawału (efekt jest niezależny od CB 1 i CB 2 )-↓ Aktywność MPO w mózgu (efekt jest niezależny od CB 1 i CB 2 )-↓ osocze HMGB1-↑ funkcje neurologiczne i koordynacja ruchowa
Okluzja MCA (4 godz.) + reperfuzja (3 dni); model udaru
3 mg/kg; bezpośrednio przed okluzją i 3 godziny po rozpoczęciu okluzji; ip
-↓ osocze HMGB1-↓ Komórki HMGB1- i MPO-dodatnie w mózgu-↓ apoptoza w mózgu-↓ aktywacja glejów w mózgu-↑ funkcje neurologiczne i koordynacja ruchowa
Mysz
okluzja MCA (4 godz.) + reperfuzja (14 dni); model udaru
3 mg/kg; przez 14, 12 lub 10 dni odpowiednio od dnia 1, 3 lub 5; ip
-↓ upośledzenie funkcji neurologicznych-↓ zaburzenia koordynacji ruchowej-↑ wskaźnik przeżycia-↓ apoptoza w mózgu-↓ liczba komórek mikrogleju (ale nie astrocytów) wyrażających HMGB1-↓ osocze HMGB1 (efekty dla CBD podawanego od 1. i 3. dnia, ale nie od 5. dnia)
Skrawki przodomózgowia zostały pozbawione tlenu i glukozy; model in vitro noworodkowego HIE
100 μmol/l
-↓ martwicza i apoptotyczna śmierć komórek-↓ ekscytotoksyczność-↓ zapalenie (efekty zależą od CB 2 i A 2 ; są niezależne od CB 1 ; ekscytotoksyczność jest również zależna od A 1 )
Niedotlenienie (90 min) i elektrokoagulacja lewej tętnicy szyjnej + okres po HI (7 dni); model noworodkowego HIE
1 mg/kg; 15 minut, 1, 3, 6, 12, 18 lub 24 godziny po HI; sc
-↓ utrata objętości półkuli ipsilateralnej-↓ zmiany histopatologiczne-↓ apoptoza-↓ astroglioza-↓ aktywacja mikrogleju (efekty dla CBD podanego do 18 h po HI) -↓ zmiany histopatologiczne-↓ aktywacja mikrogleju (efekty dla CBD podanego 24 h po HI)
5 mg/kg; 1 godzinę po okluzji, a następnie co 24 godziny przez 2 dni; IV
-↓ transaminaza alaninowa w surowicy-↓ zmiany histopatologiczne w wątrobie-↓ stres oksydacyjny i nitracyjny w wątrobie-↓ zapalenie wątroby-↓ apoptoza w wątrobie-↓ ekspresja NF-κB w wątrobie
-↓ kreatynina w surowicy-↓ zmiany histopatologiczne w nerkach-↓ stres oksydacyjny i nitracyjny w nerkach-↓ zapalenie nerek-↓ apoptoza w nerkach-↓ ekspresja NF-κB w nerkach
Zamknięcie tętnicy wątrobowej i żyły wrotnej (1 godz.) + reperfuzja (2, 6 lub 24 godz.)
3 lub 10 mg/kg; 2 godziny przed lub 90 minut po okluzji; ip
-↓ transaminazy alaninowej i asparaginianowej w surowicy (efekt jest niezależny od CB 2 )-↓ zmiany histopatologiczne w wątrobie-↓ śmierć komórek w wątrobie-↓ zapalenie wątroby-↓ ICAM-1 w wątrobie-↓ naciek neutrofili w wątrobie-↓ Aktywacja NF-κB w wątrobie-↓ Aktywacja p38 MAPK i JNK w wątrobie-↓ stres oksydacyjny i nitracyjny w wątrobie-↓ dysfunkcja mitochondriów w wątrobie
Ludzkie komórki śródbłonka tętnic wieńcowych (HCAEC) narażone na działanie wysokiej glukozy
1,5–6 μmol/l; 48 godz
-↓ cząsteczki adhezyjne ICAM-1 i VCAM-1-↓ adhezja monocytów do śródbłonka-↓ przezśródbłonkowa migracja monocytów-↓ zaburzenie funkcji bariery śródbłonkowej-↓ Aktywacja NF-κB (powyższe efekty są niezależne od CB 1 i CB 2 ) -↓ stres oksydacyjny i nitratywny
-↔ SBP, DBP, HR-↔ poziom glukozy we krwi, kontrola glikemii i wrażliwość na insulinę-↔ profil lipidowy (cholesterol HDL, cholesterol LDL, cholesterol całkowity, trójglicerydy, apolipoproteiny A i B)-↔ masa ciała-↔ adiponektyna, ↓ rezystyna, ↑ GIP we krwi
Cukrzyca indukowana streptozotocyną (model cukrzycy typu 1)
10 mg/kg (co 2 dni); przez 1, 2 lub 4 tygodnie; ip
-↔ poziom glukozy we krwi-↔ masa ciała-↓ uszkodzenie bariery krew-siatkówka-↓ śmierć komórek nerwowych w siatkówce-↓ stres oksydacyjny i nitratywny w siatkówce-↓ zapalenie siatkówki-↓ VEGF w siatkówce-↓ aktywacja p38 MAPK w siatkówce
ZDF (model cukrzycy typu 2); izolowana tętnica udowa
10 μmol/l; 2 godz
-↑ odpowiedź wazorelaksacyjna na acetylocholinę (silniejsza niż przy kontroli normoglikemicznej; działanie zależne od SOD, COX, EP 4 i CB 2 ; niezależne od śródbłonka, NO, H 2 O 2 , CB 1 , receptorów Abn-CBD i PPAR-γ)-odkrycie reakcji wazorelaksacyjnej na agonistę CB2
-↑ odpowiedź wazorelaksacyjna na acetylocholinę (ale nie na nitroprusydek sodu) w izolowanych tętnicach krezkowych (efekt zależy od COX i NO)-↔ odpowiedź wazorelaksacyjna na acetylocholinę i nitroprusydek sodu w aorcie i tętnicy udowej-↔ poziom glukozy we krwi-↓ przyrost masy ciała-↓ Peptyd C, insulina, leptyna, ICAM-1 w surowicy-↔ GLP-1, glukagon, MCP-1, polipeptyd trzustkowy, amylina, GIP, IL-6, TNF-α, peptyd YY, vWF, PAI-1 w surowicy-↑ VEGF i endotelina-1 w surowicy
Cukrzyca indukowana streptozotocyną (model cukrzycy typu 1)
1, 10 lub 20 mg/kg; przez 4 lub 11 tygodni; ip
-↔ poziom glukozy we krwi, zawartość insuliny w trzustce-↔ masa ciała-↓ dysfunkcja lewej komory-↓ stres oksydacyjny i nitratywny w sercu-↓ zapalenie i aktywacja NF-κB w sercu-↓ apoptoza i aktywacja MAPK w sercu-↓ zwłóknienie mięśnia sercowego-↓ aktywacja NF-κB, JNK, p38 i p38α MAPK w sercu-↑ aktywacja Akt w sercu
1 Efekty zaobserwowane przy co najmniej jednej z badanych dawek/stężeń; 2 pacjentów z nowotworem lub chorobą zapalną jelit; 3 agonista receptora tromboksanu; 4 chorych na raka płuc; 5 w przypadku wystąpienia znaczących skutków ubocznych dawkę zmniejszano stopniowo do 25 i 10 mg/kg. ↑/↓/↔ — wzrost/spadek/brak zmian; skróty: 4-HHE: 4-Hydroksyheksenal; 5-HT 1A, 3 : Receptory serotoninowe typu 1A, 3; Abn-CBD: nieprawidłowy kannabidiol; A 1, 2 : Receptor adenozynowy typu 1, 2; Akt: Kinaza białkowa B; BDNF: czynnik neurotroficzny pochodzenia mózgowego; BNST: jądro łoża prążkowia końcowego; BP: ciśnienie krwi; CB 1, 2 : Receptor kannabinoidowy typu 1, 2; CBF: mózgowy przepływ krwi; CK, CK-MB: Kinaza kreatynowa i jej izoenzym sercowy; CO: pojemność minutowa serca; COX: cyklooksygenaza; CRP: białko C-reaktywne; CSF: płyn mózgowo-rdzeniowy; DBP: rozkurczowe ciśnienie krwi; Sól DOCA: Sól octanu dezoksykortykosteronu; EEG: elektroencefalograficzne; EF: frakcja wyrzutowa; EJT: czas wyrzutu lewej komory; FMD: dylatacja za pośrednictwem przepływu; GDNF: czynnik neurotroficzny pochodzenia glejowego; GIP: zależny od glukozy peptyd insulinotropowy; GIP: zależny od glukozy peptyd insulinotropowy; GLP-1: glukagonopodobny peptyd-1; HDL: lipoproteina o dużej gęstości; HI: Niedotlenienie-niedokrwienie; HIE: Encefalopatia niedotlenieniowo-niedokrwienna; HMGB1: grupa o wysokiej mobilności 1; HR: tętno; ic: Wewnątrzoponowo; icv: komora mózgowa; ip: dootrzewnowo; iv: Dożylnie; ICAM-1: cząsteczka adhezji międzykomórkowej 1; IL-6: interleukina 6; JNK: N-końcowa kinaza c-Jun; LAD: lewa tętnica zstępująca przednia; LCx: lewa tętnica wieńcowa okalająca; LDH: dehydrogenaza mleczanowa; LDL: lipoproteina o małej gęstości; MAPK: kinazy białkowe aktywowane mitogenami; MBP: średnie ciśnienie krwi; MCA: tętnica środkowa mózgu; MCP-1: Białko chemoatraktacyjne monocytów-1; MDA: dialdehyd malonowy; po: Per os, ustnie; MMP2, 9: metaloproteinaza macierzy 2, 9; MPO: mieloperoksydaza; NF-кB: współczynnik jądrowy κB; PAI-1: Inhibitor aktywatora plazminogenu-1; PPAR-γ: receptor γ aktywowany przez proliferatory peroksysomów; PWV: prędkość fali pulsacyjnej; S100B: S100 białko B wiążące wapń; sc: podskórnie; SBF: przepływ krwi w skórze przedramienia; SBP: skurczowe ciśnienie krwi; SHR: szczur ze spontanicznym nadciśnieniem; SOD: dysmutaza ponadtlenkowa; SV: objętość skurczowa; TNF-α: czynnik martwicy nowotworu α; TPR: całkowity opór obwodowy; TRPV1: członek 4 podrodziny waniloidów o potencjale przejściowym; VCAM-1: białko adhezyjne komórek naczyniowych 1; VEGF: czynnik wzrostu śródbłonka naczyniowego; vWF: czynnik von Willebranda; ZDF: Zucker Diabetic Fatty Rat.
4.2. Nadciśnienie tętnicze
Nadciśnienie tętnicze wiąże się ze zmianami w układzie endokannabinoidowym (np. wzrostem AEA w osoczu), co może wskazywać na jego aktywację. Kannabinoidy podawane zwierzętom z nadciśnieniem często powodują zmianę odpowiedzi hemodynamicznej – pojawia się lub nasila faza hipotensyjna. Hamowanie FAAH może wywierać działanie hipotensyjne, które zależy od wieku zwierząt i doświadczalnego modelu nadciśnienia. Endokannabinoidy wykazały również modulujący wpływ na stres oksydacyjny i stany zapalne w nadciśnieniu, które są ważną częścią patogenezy tej choroby [ 19 , 20 , 27 , 113 , 114 ]. Biorąc pod uwagę zarówno działanie wazorelaksacyjne, modulację procesów zapalnych i oksydacyjnych, jak i metabolizm endokannabinoidów [ 23 , 27 , 30 , 31 , 71 ], można spodziewać się pewnych korzyści z ich stosowania w leczeniu nadciśnienia.
Dotychczas przeprowadzono badania na dwóch szczurzych modelach nadciśnienia (Tabela 4) — u szczurów z samoistnym nadciśnieniem (SHR; model nadciśnienia pierwotnego) i nadciśnienia indukowanego solą octanu deoksykortykosteronu (sól DOCA; model nadciśnienia wtórnego) [ 48 , 52 , 96 ]. Badania na izolowanych małych tętnicach krezkowych wykazały przeciwne działanie CBD w tych dwóch modelach. W pierwszym przypadku działanie wazorelaksacyjne CBD zostało zmniejszone, podczas gdy w drugim wzmocnione [ 48 ]. Te przeciwne efekty mogą wynikać z odmiennej patogenezy i zmian w układzie endokannabinoidowym w zastosowanych modelach nadciśnienia tętniczego [ 20 ]. Ponadto w tych dwóch modelach zaobserwowano również pewne różnice w mechanizmie działania rozszerzającego naczynia CBD (patrzTabela 4) [ 48 ]. Podobnie jak w przypadku SHR, działanie CBD rozszerzające naczynia w izolowanych tętnicach płucnych było zmniejszone u pacjentów z nadciśnieniem tętniczym [ 48 ]. Jednakże nie zaobserwowano żadnych zmian w odpowiedzi wazorelaksacyjnej na CBD w izolowanych tętnicach krezkowych osób z nadciśnieniem [ 56 ].
U przytomnych szczurów ze spontanicznym nadciśnieniem podanie ip CBD spowodowało wzrost ciśnienia krwi w pierwszych minutach po wstrzyknięciu nieco silniejszy niż w przypadku kontroli normotensyjnej [ 52 ]. Może to zatem wynikać z upośledzonego działania rozszerzającego naczynia CBD ujawnionego w SHR [ 48 ]. Jednakże efekty hemodynamiczne w SHR z rdzeniem i wagotomią (wzrost SBP i HR oraz zmniejszenie DBP) były porównywalne z grupą kontrolną [ 52 ]. U SHR znieczulonych uretanem szybkie podanie dożylne CBD wywołało silniejszy odruch Bezolda-Jarischa niż u zwierząt kontrolnych [ 52 ]. Podczas dwutygodniowego podawania CBD szczurom SHR i DOCA z solą nie zaobserwowano znaczącego wpływu na ciśnienie krwi i częstość akcji serca. Jednocześnie jednak stwierdzono spadek markerów stresu oksydacyjnego w osoczu i sercu tych zwierząt [ 96 ]. Podsumowując, dotychczasowe badania nie wykazały hipotensyjnego działania CBD w leczeniu nadciśnienia tętniczego, choć związek ten wykazuje w tej chorobie właściwości antyoksydacyjne.
Już wcześniej postulowano wysoki potencjał kardioprotekcyjny CBD (Tabela 4). Kilka badań wskazuje na korzystny wpływ na niedokrwienie/ zawał mięśnia sercowego, który uzyskano na zwierzętach doświadczalnych poprzez podwiązanie lewej tętnicy zstępującej przedniej u szczurów [ 49,105,115 ] lub lewej tętnicy wieńcowej okalającej u królików [ 116 ] . Podanie CBD przed podwiązaniem tętnicy wieńcowej i bezpośrednio przed reperfuzją zmniejszyło wielkość zawału. Dodatkowo CBD podawane przed indukcją niedokrwienia zmniejszało liczbę komorowych zaburzeń rytmu i zmniejszało agregację płytek krwi indukowaną kolagenem. Działanie antyarytmiczne CBD może wynikać z hamowania uwalniania substancji arytmogennych z płytek krwi [ 105 ]. Inne badanie wykazało, że antyarytmiczne działanie CBD na komorowe zaburzenia rytmu wywołane niedokrwieniem/reperfuzją zależy od receptorów adenozyny A 1 [ 49 ]. U królików CBD podawane przed eksperymentalnie wywołanym ostrym zawałem mięśnia sercowego zmniejszało rozmiar zawału, zwiększało przepływ krwi w obszarze z upośledzoną perfuzją, zmniejszało poziom troponiny sercowej I we krwi i zmniejszało apoptozę komórek mięśniowych. Ponadto CBD poprawiło funkcję lewej komory i chroniło przed uszkodzeniem reperfuzyjnym, co było związane ze zmniejszonym naciekiem leukocytów w sercu [ 116 ]. Przewlekłe leczenie CBD zmniejszyło również rozmiar zawału i naciek leukocytów serca w szczurzym modelu niedokrwienia i reperfuzji. Działania te były związane ze zmniejszonym poziomem interleukiny 6 w surowicy. Jednakże zmniejszenie rozmiaru zawału przez CBD zaobserwowano tylko in vivo, ale nie w izolowanych sercach. Zatem kardioprotekcyjne działanie CBD w zawale mięśnia sercowego nie wydaje się być bezpośrednie i może wynikać z jego właściwości przeciwzapalnych [ 115 ].
Kannabidiol wykazuje również potencjalne korzystne działanie w przypadku innych chorób serca (Tabela 4). Na przykład osłabia kardiotoksyczność doksorubicyny (antracyklinowego antybiotyku przeciwnowotworowego) u szczurów. Przewlekłe podawanie CBD przez cztery tygodnie zmniejszyło wywołane doksorubicyną zmiany histopatologiczne w sercu i podwyższyło w surowicy markery uszkodzenia mięśnia sercowego – kinazy kreatynowej i troponiny T. Kardioprotekcyjne działanie CBD było związane ze zmniejszeniem poziomu dialdehydu malonowego w sercu, tlenku azotu, czynnika martwicy nowotworu α ( TNF-α) i poziom jonów wapnia, zwiększony poziom zredukowanego glutationu, selenu i cynku w sercu, zmniejszona ekspresja NF-KB, indukowalnego NOS i kaspazy-3 oraz zwiększona ekspresja surwiwiny [ 117 ]. W innym badaniu leczenie CBD przez pięć dni poprawiło dysfunkcję serca wywołaną doksorubicyną, zmniejszyło aktywność kinazy kreatynowej i dehydrogenazy mleczanowej w surowicy (markery uszkodzenia serca). Podobnie jak w poprzednim badaniu, leczenie CBD znacznie zmniejszyło stres oksydacyjny i nitratywny oraz śmierć komórek w kardiomiopatii wywołanej doksorubicyną. Ponadto CBD wzmocniło upośledzoną funkcję mitochondriów serca i biogenezę w tej patologii [ 118 ]. W modelu eksperymentalnego autoimmunologicznego zapalenia mięśnia sercowego u myszy zastrzyki z CBD poprawiły właściwości skurczowe i rozkurczowe lewej komory, jednocześnie zmniejszając jej przebudowę zwłóknieniową, stan zapalny, martwicę i naciek jednojądrowy. Badania biochemiczne potwierdziły złagodzenie stanu zapalnego związane ze zmniejszoną ekspresją cytokin prozapalnych (interleukiny 6 i 1β oraz interferonem-γ) oraz poziomem sercowego 4-hydroksynonenalu i 3-nitrotyrozyny (odpowiednio markerów stresu oksydacyjnego i nitracyjnego) [ 119 ]. Właściwości kardioprotekcyjne CBD wykazano także w zwierzęcym modelu cukrzycy (patrz poniżej).
4.4. Udar mózgu, noworodkowa encefalopatia niedotlenieniowo-niedokrwienna, zapalenie mózgu związane z sepsą
Właściwości neuroprotekcyjne CBD wykazano w szerokiej gamie modeli zwierzęcych zaburzeń neurologicznych, w tym epilepsji, choroby Alzheimera, choroby Huntingtona, choroby Parkinsona i stwardnienia rozsianego [ 11 , 17 , 30 , 66 ]. Istnieją również pewne dowody (np.Tabela 4) pod kątem korzystnego wpływu CBD na zaburzenia mózgu związane z niedotlenieniem i/lub niedokrwieniem, takie jak udar i noworodkowa encefalopatia niedotlenieniowo-niedokrwienna (HIE).
Wpływ kannabidiolu na udar niedokrwienny mózgu badano głównie na myszach i szczurach z niedrożnością tętnicy środkowej mózgu (Tabela 4). W tym modelu udaru CBD podawane zarówno przed, jak i/lub po niedokrwieniu zmniejszało objętość zawału [ 50 , 53 , 120 , 121 , 122 ] (ale nie u nowonarodzonych szczurów [ 123 ]) i poprawiało upośledzone funkcje neurologiczne i/lub neurobehawioralne [ 50 , 120 , 121 , 122 , 123 , 124 ]. CBD zwiększa mózgowy przepływ krwi podczas okluzji [ 50,53 ] , co jest zgodne z metaanalizą Sultana i in . [ 24 ], które wskazywały na zwiększone CBF w mysich modelach udaru po podaniu CBD. Ponadto jedno badanie wykazało, że CBD hamowało również spadek CBF z powodu niewydolności mikrokrążenia mózgowego przez 1 godzinę po reperfuzji [ 50 ]. Wpływ CBD na objętość zawału i CBF odbywał się, przynajmniej częściowo, za pośrednictwem receptorów serotoninowych 5-HT 1A i nie był zależny od receptorów kannabinoidowych i receptorów waniloidowych TRPV1 [ 50 , 53 , 121 ]. Co ważne, powtarzane leczenie CBD przez 14 dni przed okluzją nie wykazało rozwoju tolerancji na jego właściwości neuroprotekcyjne [ 50 ]. Wykazano, że CBD wykazuje właściwości neuroprotekcyjne przy podawaniu nawet po dłuższym czasie od niedokrwienia mózgu. Zatem powtarzane leczenie CBD najpóźniej od pierwszego lub trzeciego dnia po indukcji udaru poprawiło deficyty funkcjonalne, wskaźniki przeżycia i uszkodzenia niedokrwienne [ 124 ]. U myszoskoczków i szczurów narażonych na niedokrwienie mózgu wywołane obustronnym zamknięciem tętnicy szyjnej wykazano działanie ochronne CBD przed neurodegeneracją hipokampa i zaburzeniami funkcji poznawczych lub nadpobudliwością ruchową [ 125,126 ]. Neuroprotekcyjne działanie CBD w zwierzęcych modelach udaru było związane ze zmniejszoną ekscytotoksycznością [ 123 ] , aktywacją glejów [ 121,123,124,126 ] , upośledzeniem metabolizmu neuronalnego [ 123 ] i apoptozą [ 121,123,124 ] . CBD zmniejszyło także zapalenie układu nerwowego wywołane udarem, ponieważ zmniejszyło liczbę komórek dodatnich pod względem mieloperoksydazy (neutrofili) [ 50,121 ] i zmniejszyło ekspresję czynników zapalnych, takich jak receptor TNF-α 1 i NF-KB w mózgu [ 122] Ponadto CBD może łagodzić urazy po niedokrwieniu poprzez hamowanie białka grupy 1 o wysokiej ruchliwości (HMGB1). To niehistonowe białko wiążące DNA jest masowo uwalniane do przestrzeni zewnątrzkomórkowej z komórek zapalnych i martwiczych po niedokrwieniu i indukuje ekspresję genów związanych z zapaleniem układu nerwowego i aktywacją mikrogleju. Rzeczywiście, leczenie CBD zmniejszyło poziom HMGB1 w osoczu i liczbę komórek HMGB1-dodatnich w mózgu myszy poddanych ogniskowemu niedokrwieniu mózgu [ 121,124 ] . Należy zauważyć, że w niedokrwieniu mózgu CBD wywołało pewne korzystne skutki w postaci zależnej od dawki krzywej dzwonowej. Zatem zapobieganie spłaszczeniu elektroencefalograficznemu było największe przy dawce 5 mg/kg CBD (1,25–20 mg/kg) [ 125 ] i zmniejszeniu objętości zawału przy dawce 1 mg/kg CBD (0,1–10 mg/kg) [ 53 ].
Najczęstszą przyczyną uszkodzeń mózgu u noworodków jest okołoporodowa encefalopatia niedotlenieniowo-niedokrwienna spowodowana uduszeniem. Kannabidiol jest uważany za obiecujący środek neuroprotekcyjny w leczeniu niedotlenienia-niedokrwienia noworodków i jak wspomniano powyżej, w Unii Europejskiej związek ten ma nawet status leku sierocego w leczeniu zamartwicy okołoporodowej. Najwięcej dowodów na ochronne działanie CBD w HIE opiera się na badaniach na nowonarodzonych prosiętach poddanych niedotlenieniu i niedokrwieniu [ 54,127,128,129,130,131 ] (Tabela 4). Korzystny wpływ CBD na eksperymentalny HIE noworodków obejmuje złagodzenie zmniejszonej aktywności mózgu [ 54,127,128,129,130,131 ] , upośledzenie metabolizmu neuronalnego [ 127,128,129,131 ] , ekscytotoksyczność [ 54,129 ] , zmiany histopatologiczne w mózgu [ 127 , 128 ], martwica i/lub apoptoza neuronów [ 54 , 129 , 130 , 131 ], aktywacja astrogleju i/lub mikrogleju [ 131 ], zapalenie nerwów [ 54 , 128 , 129 , 130 , 131 ] i stres oksydacyjny w mózg [ 54 , 130 , 131 ]. CBD może również zmniejszać odległe zapalne uszkodzenia płuc związane z uszkodzeniem mózgu wywołanym niedotlenieniem i niedokrwieniem [ 130 ]. Ponadto CBD może zmniejszać spadek ciśnienia krwi wywołany niedotlenieniem i niedokrwieniem [ 54,127,129,130,131 ] . Jednakże wysoka dawka CBD (50 i 25 mg/kg; najczęstsza dawka w poprzednich badaniach wynosiła 1 mg/kg) wywołała znaczne niedociśnienie u niektórych prosiąt (CBD 50 mg/kg u dwóch z czterech prosiąt i CBD 25 mg/kg kg u jednego na cztery prosięta). Ponadto u jednego prosiaka doszło do śmiertelnego zatrzymania akcji serca po wlewie CBD w dawce 50 mg/kg [ 132 ]. Dlatego należy zachować ostrożność podczas leczenia dużymi dawkami CBD ze względu na możliwość wystąpienia działań niepożądanych ze strony układu sercowo-naczyniowego. Korzystne działanie CBD wykazano także w mysich [ 133 ] i szczurzych modelach [ 134 , 135 ] noworodkowego HIE (Tabela 4). Warto zauważyć, że CBD wykazuje szersze okno czasu terapeutycznego niż zgłaszane w przypadku hipotermii i innych terapii neuroprotekcyjnych [ 133 ]. Ochronne działanie CBD w przypadku uszkodzenia mózgu wywołanego niedotlenieniem i niedokrwieniem zachodzi, przynajmniej częściowo, poprzez receptory CB 2 i 5-HT 1A [ 54 , 130 ]. Badania na skrawkach przodomózgowia nowonarodzonych myszy poddanych pozbawieniu tlenu i glukozy wykazały, że oprócz receptorów CB2 , receptory adenozynowe (głównie A2 ) mogą również pośredniczyć w neuroprotekcyjnym działaniu CBD [ 136 ]. Jednakże niektórzy autorzy podali, że CBD ( w niskich lub wysokich dawkach ) nie wykazywało żadnych właściwości neuroprotekcyjnych w modelach eksperymentalnych noworodkowego HIE [ 137 ]. Niemniej jednak CBD może zwiększyć ochronne działanie hipotermii, co jest złotym standardem w leczeniu niemowląt z HIE [ 129 , 131 ].
Przeciwzapalne i stabilizujące naczynia działanie CBD wykazano także w przypadku mysiego zapalenia mózgu wywołanego podaniem lipopolisacharydu (LPS) (Tabela 4). LPS powodował rozszerzenie naczyń tętniczych i żylnych, zwiększoną marginalizację leukocytów, zwiększoną ekspresję prozapalnych TNF-α i COX-2, wyższy poziom markerów stresu oksydacyjnego (dialdehydu malonowego i 4-hydroksynonenalu) oraz zaburzenie bariery krew-mózg. Leczenie CBD złagodziło prawie wszystkie zmiany wywołane LPS (z wyjątkiem markerów stresu oksydacyjnego), a dodatkowo zmniejszyło indukowaną ekspresję NOS. Zatem CBD może stanowić opcję leczenia zapalenia mózgu i encefalopatii związanego z sepsą [ 138 ].
4,5. Niedokrwienie/uszkodzenie reperfuzyjne nerek i wątroby
Wykazano, że kannabidiol chroni przed uszkodzeniem nerek i wątroby w wyniku niedokrwienia/reperfuzji (Tabela 4). Tego typu uszkodzenia mogą wystąpić podczas wstrząsu oraz operacji lub przeszczepienia tych narządów. W szczurzym modelu uszkodzenia nerek z powodu niedokrwienia/reperfuzji, CBD znacząco zmniejszyło zmiany histopatologiczne w nerkach i obniżyło poziom kreatyniny w surowicy (marker czynności nerek). Nefroprotekcyjne działanie CBD było związane ze złagodzeniem stresu oksydacyjnego i nitracyjnego wywołanego niedokrwieniem/reperfuzją, stanem zapalnym i apoptozą [ 139 ]. Podobne działanie ochronne uzyskano u gryzoni poddanych niedokrwieniu/reperfuzji wątroby. Zatem CBD zmniejszyło transaminazy w surowicy (markery uszkodzenia wątroby) i zmiany histopatologiczne, śmierć komórek, stres oksydacyjny i nitracyjny oraz zapalenie wątroby [ 140,141 ] . Wykazano, że mechanizm tego działania hepatoprotekcyjnego może obejmować osłabioną aktywację NF-κB, p38 MAPK i JNK przez CBD [ 141 ]. Badania in vitro na komórkach sinusoidalnych ludzkiej wątroby wykazały, że CBD może osłabiać indukowaną przez TNF-α ekspresję cząsteczek adhezyjnych (ICAM-1 i VCAM-1) oraz adhezję komórek wielojądrzastych do komórek sinusoidalnych wątroby. Odpowiada to indukowanemu przez CBD zmniejszeniu ekspresji ICAM-1 i naciekowi neutrofili w wątrobie myszy poddanej uszkodzeniu niedokrwiennemu/reperfuzyjnemu [ 141 ]. Wydaje się, że hepatoprotekcyjne działanie CBD nie jest zależne od receptorów kannabinoidowych, ponieważ nie zostało osłabione przez antagonistów CB 1 i CB 2 in vitro i nadal występowało u myszy z nokautem CB 2 [ 141 ]. Podsumowując, CBD ma ogromny potencjał terapeutyczny w zapobieganiu i łagodzeniu uszkodzeń niedokrwiennych/reperfuzyjnych różnych narządów, takich jak serce, mózg, nerki i wątroba.
4.6. Powikłania sercowo-naczyniowe cukrzycy
Cukrzyca powoduje wiele powikłań związanych z sercem i naczyniami krwionośnymi, takich jak miażdżyca, retinopatia lub kardiomiopatia, które są związane z dysfunkcją śródbłonka naczyń, zwiększonym stanem zapalnym i stresem oksydacyjnym. CBD nie wpływa na poziom glukozy we krwi u zwierząt chorych na cukrzycę [ 142 , 143 , 144 ] i ludzi [ 145 ]. Ponadto u pacjentów z cukrzycą typu 2 CBD nie wpływało na kontrolę glikemii, wrażliwość na insulinę, profil lipidowy, masę ciała i parametry hemodynamiczne [ 145 ]. Jednakże, ze względu na swoje właściwości przeciwutleniające, przeciwzapalne oraz naczyniowe, kardiologiczne i neuroprotekcyjne, CBD może łagodzić powikłania sercowo-naczyniowe cukrzycy (Tabela 4).
Zakłócenie funkcji i integralności śródbłonka jest niezbędne dla rozwoju różnych powikłań cukrzycy. W ludzkich komórkach śródbłonka tętnic wieńcowych narażonych na działanie wysokiej glukozy wykazano zwiększone wytwarzanie mitochondrialnych nadtlenków, tworzenie 3-nitrotyrozyny, aktywację NF-κB, indukowalną ekspresję NOS i cząsteczek adhezyjnych, przezśródbłonkową migrację monocytów i ich adhezję do śródbłonka. Wstępne leczenie CBD złagodziło wszystkie te negatywne skutki. Ponadto poprawił także zaburzenia funkcji bariery śródbłonkowej wywołane wysokim poziomem glukozy. Ochronne działanie CBD na komórki śródbłonka było niezależne od CB 1 i CB 2 [ 146 ].
Wpływ CBD na funkcję naczyń krwionośnych badano u szczurów Zucker Diabetic Fat Rats (ZDF) — model cukrzycy typu 2. Inkubacja in vitro z CBD wzmocniła działanie rozszerzające naczynia krwionośne acetylocholiny w izolowanej aorcie i tętnicy udowej, a efekt ten był silniejszy w porównaniu ze zwierzętami kontrolnymi z normoglikemią [ 57,147 ] . Badania badające mechanizm działania w tętnicach udowych wykazały, że CBD aktywuje cyklooksygenazę, a następnie indukuje wytwarzanie związków aktywujących rozszerzające naczynia receptory prostanoidowe EP 4 . Co więcej, działanie CBD było zależne od dysmutazy ponadtlenkowej i receptorów CB2 . Co ciekawe, w tętnicach udowych ZDF CBD odkryło działanie rozszerzające naczynia krwionośne agonisty receptora CB 2 HU308 (związek ten bez obecności CBD nie wykazywał działania rozszerzającego naczynia) [ 57 ]. Kannabidiol może również poprawiać rozluźnienie naczyń u szczurów z cukrzycą po leczeniu in vivo. Zatem wielokrotne podawanie CBD przez siedem dni znacząco zwiększało rozszerzenie naczyń do acetylocholiny w izolowanych tętnicach krezkowych (ale nie w aorcie i tętnicach udowych), a efekt ten był wrażliwy na hamowanie COX i NOS. Ponadto CBD zmniejszyło niektóre biomarkery metaboliczne i sercowo-naczyniowe w surowicy (patrzTabela 4). Jednakże, co ciekawe, zwiększył poziom krążącej endoteliny 1, co jest niespójne z poprawą funkcji naczyń [ 144 ]. Warto zauważyć, że u ludzi chorych na cukrzycę typu 2 CBD nie tylko nie wykazywało zwiększonych właściwości rozszerzających naczynia (w izolowanych tętnicach płucnych) [ 48 ], ale reakcje wazorelaksacyjne CBD były osłabione (w tętnicach krezkowych) [ 144 ].
Leczenie kannabidiolem może być korzystne w przypadku retinopatii cukrzycowej, która charakteryzuje się zwiększoną przepuszczalnością naczyń i neurotoksycznością. U szczurów z cukrzycą indukowaną streptozotocyną (model cukrzycy typu 1) przewlekłe podawanie CBD poprawiło funkcję bariery krew-siatkówka, zmniejszyło stres oksydacyjny i nitratywny, obniżyło poziom TNF-α i ICAM-1 oraz zapobiegło śmierci komórek nerwowych w siatkówce. Ponadto CBD obniżyło w siatkówce poziom czynnika wzrostu śródbłonka naczyniowego (VEGF), co powiązano z rozkładem bariery krew-siatkówka [ 142 ]. I odwrotnie, CBD zwiększyło krążący VEGF u szczurów ZDF, zatem wpływ CBD na tego mediatora wymaga dalszych badań [ 144 ]. Właściwości ochronne CBD w siatkówce cukrzycowej mogą wynikać z hamowania aktywacji p38 MAPK. Ta kinaza białkowa jest dalszym celem stresu oksydacyjnego i cytokin prozapalnych, a jej aktywacja może zwiększać przepuszczalność naczyń i śmierć komórek, kluczowe elementy patogenezy retinopatii cukrzycowej [ 142 ].
Kolejnym powikłaniem cukrzycy jest kardiomiopatia, charakteryzująca się dysfunkcją rozkurczową, a następnie skurczową lewej komory. Patogeneza kardiomiopatii cukrzycowej jest złożona i obejmuje stres oksydacyjny/nitracyjny, stan zapalny, zwłóknienie serca i śmierć kardiomiocytów. U myszy z cukrzycą indukowaną streptozotocyną, przewlekle podawany CBD łagodzi wszystkie te zmiany poprzez hamowanie szlaków prozapalnych i śmierci komórkowej (NF-κB, p38 i p38α MAPK, JNK) oraz wzmacnianie szlaku sygnalizacyjnego prosuvival (Akt) [143 ] . Podobnie w ludzkich kardiomiocytach CBD eliminowało niekorzystne skutki hiperglikemii poprzez zmniejszenie stresu oksydacyjnego i nitracyjnego, aktywację NF-κB i apoptozę komórek [ 143 ].
5. Wpływ nieprawidłowego kannabidiolu na układ sercowo-naczyniowy
Nieprawidłowy-kannabidiol (struktura chemicznaRysunek 4) jest syntetycznym regioizomerem kanabidiolu, który we wstępnych badaniach nie wykazywał jawnych skutków behawioralnych [ 51 , 148 , 149 ]. Abn -CBD powoduje niedociśnienie u znieczulonych psów [ 148 ] i myszy [ 51,150 ] po podaniu ogólnoustrojowym oraz rozszerzenie naczyń w izolowanych myszach i łożysku krezkowym szczura [ 51 ] , tętnicach krezkowych szczura [ 150,151,152 ] i szczurze [ 153 ], królika [ 154 ] i ludzkie tętnice płucne [ 155 ]. Efekty te nie są zależne od receptorów kannabinoidowych. Działanie wazorelaksacyjne Abn-CBD zachodzi hipotetycznie poprzez niezidentyfikowany receptor śródbłonkowy zwany receptorem Abn-CBD lub śródbłonkowy receptor kannabinoidowy i jest hamowane przez CBD i O-1918, domniemanych antagonistów tego receptora [ 51 , 71 , 73 ]. Jednak w oparciu o obecną wiedzę istnienie specyficznego śródbłonkowego receptora dla Abn-CBD jest dość kontrowersyjne. Abn-CBD jest agonistą GPR18, a O-1918 i CBD służą odpowiednio jako antagonista lub częściowy agonista/antagonista tego receptora [ 73 , 156 ]. Dlatego niektórzy autorzy sugerują, że proponowanym śródbłonkowym receptorem kannabinoidowym może być GPR18. Jednak nie wszystkie obserwacje eksperymentalne potwierdzają to jednoznacznie [ 73 ]. Ponadto niezależna od GPR18 aktywacja kanałów K + (BK Ca ) o wysokim przewodnictwie aktywowanych Ca 2+ może przyczyniać się do rozszerzającego naczynia działania Abn-CBD [ 157 ]. Abn-CBD może również obniżać ciśnienie krwi po podaniu wewnątrz RVLM poprzez aktywację GPR18, co prowadzi do hamowania sympatotropii [ 158 ]. Ponadto związek ten jest agonistą GPR55, jednakże receptor ten nie pośredniczy w odpowiedzi wazorelaksacyjnej na Abn-CBD [ 150 ].
Rysunek 4
Struktura chemiczna nieprawidłowego kannabidiolu.
Przewlekle podawany Abn-CBD wywierał korzystny wpływ na układ sercowo-naczyniowy u szczurów poprzez obniżenie średniego ciśnienia krwi, zwiększenie stężenia adiponektyny w osoczu i sercu, zwiększenie dostępności NO w naczyniach, poprawę funkcji lewej komory czy zmniejszenie reaktywnych form tlenu w sercu [ 159 ] Abn-CBD wykazało także działanie kardioprotekcyjne u szczurów z cukrzycą wywołaną streptozocyną. Łagodził dysfunkcję serca (ale nie przerost serca), dominację nerwu błędnego, stres oksydacyjny mięśnia sercowego oraz zmniejszał poziom NO i adiponektyny w sercu i/lub krążeniu u szczurów z cukrzycą [ 160 ]. Wszystkie te korzystne efekty zostały zniesione przez O-1918, wskazując na rolę GPR18 w działaniu ochronnym Abn-CBD w układzie sercowo-naczyniowym [ 159 , 160 ]. Kardioprotekcji obserwowanej u szczurów z cukrzycą nie towarzyszyła poprawa kontroli glikemii – Abn-CBD nie wpływało na poziom glukozy i insuliny we krwi [ 160 ]. Jednakże w innych badaniach związek ten wykazuje potencjał przeciwcukrzycowy poprzez aktywację GPR55 [ 161 , 162 , 163 , 164 ]. Abn-CBD ma również właściwości neuroprotekcyjne, ponieważ zmniejsza objętość zawału równie silnie, jak CBD w mysim modelu udaru [ 53 ]. Podsumowując, zarówno CBD, jak i jego syntetyczny analog Abn-CBD ma właściwości rozszerzające naczynia krwionośne (jednocześnie CBD hamuje działanie wazorelaksacyjne Abn-CBD) i wykazuje potencjalnie korzystne działanie na układ sercowo-naczyniowy, jednak poprzez różne (nawet częściowo przeciwstawne) mechanizmy . Tę pozorną sprzeczność można wytłumaczyć złożonym i wielokierunkowym mechanizmem działania CBD, np. (1) działanie CBD rozszerzające naczynia następuje poprzez bezpośrednią lub pośrednią aktywację różnych receptorów (w zależności od łożyska naczyniowego), w tym CB 1 , TRPV1, EP 4 , IP i PPAR-γ [ 48 , 56 , 106 ]; (2) CBD może działać zarówno jako antagonista, jak i częściowy agonista GPR18 [ 156 ]; oraz (3) CBD bezpośrednio antagonizuje GPR55 [ 11 ], ale może potencjalnie aktywować ten receptor pośrednio poprzez hamowanie degradacji endokannabinoidów [ 27 ].
6. Wnioski
W artykule dokonano przeglądu wpływu kannabidiolu, nieodurzającego składnika konopi indyjskich o szerokim potencjale terapeutycznym i dobrym profilu bezpieczeństwa, na układ sercowo-naczyniowy w warunkach fizjologicznych i patologicznych (podsumowane w:Rysunek 5). CBD może wpływać na układ sercowo-naczyniowy poprzez różne mechanizmy bezpośrednie i pośrednie. Szczegółowe określenie wpływu CBD na układ sercowo-naczyniowy jest istotne ze względu na stale rosnące wykorzystanie tego związku w celach terapeutycznych (w tym samoleczenia) lub rekreacyjnych. Jednakże, z kilkoma wyjątkami, wpływ CBD na układ sercowo-naczyniowy w warunkach fizjologicznych wydaje się znikomy, co potwierdza dobry profil bezpieczeństwa tego kannabinoidu. Z drugiej strony rozważane jest potencjalne zastosowanie CBD w leczeniu zaburzeń sercowo-naczyniowych. W eksperymentalnych stanach patologicznych, takich jak nadciśnienie, choroby serca, udar, niedotlenienie-niedokrwienie noworodków, cukrzyca lub niedokrwienie/uszkodzenie reperfuzyjne wątroby i nerek, ochronne działanie CBD związane z jego działaniem przeciwzapalnym, przeciwutleniającym, przeciwapoptotycznym, naczynioprotekcyjnym, kardioprotekcyjnym lub neuroprotekcyjnym efekty często się ujawniają. Pomimo właściwości rozszerzających naczynia krwionośne, w zwierzęcych modelach nadciśnienia nie wykazano, że CBD wykazuje działanie hipotensyjne. Jednakże związek ten może obniżyć wywołany stresem wzrost ciśnienia krwi zarówno u ludzi, jak i zwierząt. Niemniej jednak należy podkreślić, że prawie nie przeprowadzono badań klinicznych CBD w chorobach układu sercowo-naczyniowego, w związku z czym jego potencjał terapeutyczny nie przekłada się na praktykę kliniczną. Dalsze badania, szczególnie kliniczne, uzasadniają zalecenie stosowania CBD w leczeniu zaburzeń sercowo-naczyniowych.
Układ sercowo-naczyniowy wydaje się być celem terapeutycznym dla kannabidiolu (CBD), co wynika z kilku obszarów badań:
Efekty na Naczynia Krwionośne:
CBD wykazuje zdolność do bezpośredniego rozszerzania naczyń krwionośnych, zarówno ostrych, jak i czasowych, co może wpływać korzystnie na przepływ krwi.
W warunkach laboratoryjnych CBD poprawia zdolność naczyń do rozszerzania się w odpowiedzi na bodźce zależne od śródbłonka, co może być korzystne w przypadku dysfunkcji śródbłonka.
Wpływ na Ciśnienie Krwi i Rytm Serca:
Leczenie CBD nie wydaje się wpływać na spoczynkowe ciśnienie krwi ani częstość akcji serca, co sugeruje, że może być stosunkowo bezpieczne w tym kontekście.
Ochrona Przed Stresem Sercowo-Naczyniowym:
CBD redukuje odpowiedzi układu sercowo-naczyniowego na różne rodzaje stresu, co może pomóc w ochronie serca w sytuacjach stresowych.
Ochrona Przed Uszkodzeniami Serca:
CBD wykazuje ochronne działanie w przypadku niedokrwienia i reperfuzji serca, a także w przypadku dysfunkcji serca związanego z cukrzycą, co sugeruje potencjalne zastosowanie w leczeniu tych schorzeń.
Ochrona Przed Uszkodzeniami Mózgu:
CBD może także chronić przed uszkodzeniami w przypadku udaru mózgu, poprzez utrzymanie przepływu krwi i redukcję przepuszczalności bariery krew-mózg.
Redukcja Zmian Związanych z Cukrzycą:
CBD wydaje się redukować szkodliwe zmiany związane z cukrzycą, takie jak zwiększona przepuszczalność naczyń krwionośnych.
Badania te sugerują, że CBD może mieć pozytywny wpływ na układ sercowo-naczyniowy poprzez różnorodne mechanizmy, w tym poprawę funkcji naczyń krwionośnych, ochronę przed stresem i uszkodzeniami, a także redukcję negatywnych efektów związanego z cukrzycą.
Kannabidiol (CBD) wykazuje korzystne efekty w zakresie różnorodnych schorzeń, takich jak cukrzyca, choroba Huntingtona, nowotwory i wrzody jelita grubego. Narastające dowody sugerują również, że CBD jest korzystny dla układu sercowo-naczyniowego. CBD wywiera bezpośrednie działania na izolowane tętnice, powodując zarówno ostre, jak i czasowo zależne rozszerzenie naczyń. Inkubacja in vitro z CBD wzmacnia odpowiedzi rozszerzające naczynia w modelach zwierzęcych z upośledzonym naczyniowym rozszerzaniem zależnym od śródbłonka. CBD chroni przed uszkodzeniem naczyniowym wywołanym przez środowisko o wysokiej zawartości glukozy, stan zapalny lub indukowanie cukrzycy typu 2 u zwierząt, a także redukuje nadmierną przepuszczalność naczyniową związaną z takimi środowiskami. Powszechnym motywem w tych badaniach jest przeciwzapalne i przeciutleniające działanie CBD. W sercu, leczenie CBD in vivo chroni przed uszkodzeniem przez niedokrwienie-reperfuzję i przed kardiomiopatią związaną z cukrzycą. Podobnie, w innym modelu niedokrwienia-reperfuzji, CBD wykazuje zdolność do zmniejszania rozmiaru zawału i zwiększania przepływu krwi w modelach zwierzęcych udaru, czułych na antagonizm receptora 5HT1A. Chociaż ostre lub chroniczne leczenie CBD wydaje się mieć niewielki wpływ na hemodynamikę, CBD redukuje odpowiedź sercowo-naczyniową na modele stresu, zastosowane zarówno ogólnoustrojowo, jak i do mózgu, zahamowane przez antagonistę receptora 5HT1A. W krwi CBD wpływa na przeżycie i śmierć białych krwinek, migrację białych krwinek i agregację płytek krwi. Podsumowując, te dane przedkliniczne zdają się potwierdzać pozytywną rolę leczenia CBD dla serca oraz dla naczyń obwodowych i mózgowych.
Kannabidiol (CBD) to obfity, niepsychoaktywny, pochodzący z rośliny kannabinoid (fitokannabinoid), którego stereochemia została po raz pierwszy opisana w 1963 roku przez Mechoulama i jego kolegów [1]. Izolacja struktury chemicznej CBD ujawniła, że jest to klasyczny kannabinoid blisko spokrewniony z kannabinolem i Δ−9-tetrahydrokannabinolem (THC). Od czasu jego izolacji opracowano szereg analogów syntetycznych opartych na klasycznej strukturze dibenzopyranu kannabinoidów, w tym nietypowego CBD (Abn-CBD), O-1918 i O-1602 [2, 3]. CBD ma różnorodną farmakologię, która została szczegółowo omówiona w innych miejscach [4]. W skrócie, CBD wykazuje antagonizm wobec klasycznych receptorów kannabinoidowych 1 (CB1) i 2 (CB2) w zakresie niskich nanomoli, ale wykazuje działania agonistyczne/inwersyjne agonisty w stężeniach mikromolowych. Inne miejsca receptorowe zaangażowane w działanie CBD obejmują sieroce receptory sprzężone z białkiem G GPR55, putatywny receptor Abn-CBD, receptor transient receptor potential vanilloid 1 (TRPV1), receptory α1-adrenergiczne, receptory opioidowe µ i receptory 5HT1A [4]. Wykazano również, że CBD aktywuje i wywołuje odpowiedzi fizjologiczne za pośrednictwem receptora gamma aktywowanego przez proliferatory peroksysomów (PPARγ) [5–7]. Oprócz bogatej farmakologii, sugeruje się, że CBD ma potencjał terapeutyczny w szerokim zakresie zaburzeń, w tym stanów zapalnych, stresu oksydacyjnego, nowotworów, cukrzycy, zaburzeń przewodu pokarmowego, zaburzeń neurodegeneracyjnych i nocicepcji [8–12]. Dowody wskazują również, że CBD wywiera pozytywne efekty w naczyniach krwionośnych. Celem tej recenzji jest zbadanie tych dowodów i ustalenie, czy układ sercowo-naczyniowy jest potencjalnym celem terapeutycznym dla CBD. Ostatnia recenzja dotycząca bezpieczeństwa i skutków ubocznych CBD doszła do wniosku, że CBD wydaje się być dobrze tolerowany w wysokich dawkach i przy stosowaniu chronicznym u ludzi [13], co sugeruje jego potencjał bezpiecznego stosowania w klinice.
Ostre efekty naczyniowe związków kannabinoidowych zostały dobrze zbadane w różnych modelach. W różnych modelach in vivo i in vitro wykazano, że fitokannabinoidy i endokannabinoidy (kannabinoidy endogenne) powodują rozszerzenie naczyń. Jednakże potencjał, skuteczność i mechanizmy działania często się różnią. Na przykład wcześniejsze badania na naczyniach mózgowych królika wykazały, że THC i endokannabinoid anandamid (AEA) wywołujące rozszerzenie naczyń zależne było od aktywności cyklooksygenazy (COX) [14]. Później Randall i inni [15] wykazali, że AEA indukował rozszerzenie naczyń w perfundowanym łóżku krezkowym szczura, co zostało zahamowane przez antagonizm receptora CB1 i hamowanie hiperpolaryzacji potasu. Efekty rozszerzające naczynia AEA w tętnicach szczura są również zależne od rozmiaru naczynia; maksymalna odpowiedź na AEA jest większa w małych naczyniach oporu i obejmuje składnik zależny od śródbłonka, który nie występuje w większych tętnicach [16]. W aortach szczura odpowiedź rozszerzająca naczynia na AEA nie jest wrażliwa na antagonizm CB1 ani na desensytyzację TRPV1, ale jest wrażliwa na inhibicję białka Gi/o przy użyciu toksyny krztuśca (PTX) [17]. Dodatkowe różnice w efektach kannabinoidów można zaobserwować, porównując ten sam łóżko naczyniowe różnych gatunków. W aortach króliczych AEA wywołuje większe maksymalne rozszerzenie niż zaobserwowano w aortach szczura za pośrednictwem ścieżki wrażliwej na SR141716A (1 µm), która zależy od śródbłonka [18]. Wykazano również, że odpowiedzi na kannabinoidy zależą od związku kannabinoidowego użytego. Na przykład endokannabinoidy AEA i N-arachidonoyl-dopamina (NADA) wywołują podobne stopnie rozszerzenia naczyń w aortach szczura, ale przez różne mechanizmy [17]. Te badania podkreślają złożoność ostrych działań rozszerzających naczynia kannabinoidów na układzie krwionośnym (dla pełnej recenzji, zobacz [19]).
Oprócz bezpośrednich efektów naczyniowych kannabinoidów, wiele badań wskazuje, że endokannabinoidy są mediatorami zawału mięśnia sercowego i uszkodzenia niedokrwienia/reperfuzji, czynników ryzyka układu sercowo-naczyniowego oraz miażdżycy [20–22]. Potencjalne terapeutyczne zastosowania kannabinoidów innych niż CBD w chorobach sercowo-naczyniowych, takich jak ochrona serca, udar, zaburzenia rytmu serca i miażdżyca, zostały omówione gdzie indziej [22–24].
Dotychczasowe badania dotyczące wpływu kannabinoidów na układ krwionośny skupiały się głównie na reakcjach na endokannabinoidy, THC oraz syntetyczne ligandy, z tylko ograniczoną liczbą przeprowadzonych badań z użyciem CBD. Niemniej jednak efekty analogu CBD, jakim jest Abn-CBD, zostały zbadane. Jarai i inni [25] pokazali, że Abn-CBD powoduje hipotensję zarówno u myszy CB1+/+/CB2+/+, jak i u myszy CB1−/−/CB2−/−. Efekty Abn-CBD były hamowane przez wysokie stężenia SR141716A, denudację śródbłonka i CBD. W tym artykule pokazano, że CBD antagonizuje efekty rozszerzające naczynia Abn-CBD i AEA. Begg i inni [26] wykazali w komórkach śródbłonka ludzkiego pępkowego (HUVECs), że Abn-CBD powoduje hiperpolaryzację poprzez PTX-czułą aktywację dużych kanałów potasowych aktywowanych wapniem (BKCa). Podobnie, w izolowanych tętnicach krezkowych szczura Abn-CBD powoduje rozszerzenie naczyń zależne od śródbłonka, szlaku wrażliwego na SR141716A oraz hiperpolaryzacji potasowej poprzez kanały potasowe aktywowane wapniem o dużym, pośrednim i małym przewodnictwie (BKCa/IKCa/SKCa) [27]. Co ciekawe, wcześniejsze badania raportowały szlak niezależny od śródbłonka, który obejmował modyfikację kanałów Ca2+ przez Abn-CBD. Wyniki składające się z komponentów Abn-CBD-indukowanego rozszerzenia naczyń zależnego i niezależnego od śródbłonka są zgodne z podobnym badaniem tego samego roku, które wykazało, że w tętnicach krezkowych szczura rozszerzenie naczyń indukowane przez Abn-CBD było hamowane przez inkubację PTX oraz inkubację z innym analogiem CBD, O-1918, i rzeczywiście było zależne od śródbłonka [28]. Ostatnio pokazano również, że Abn-CBD powoduje rozszerzenie naczyń w tętnicy płucnej u ludzi poprzez podobne mechanizmy [29]. Te wyniki potwierdzają obecność związanej z śródbłonkiem receptora sprzężonego z białkiem Gi/o, który powoduje rozszerzenie naczyń poprzez hiperpolaryzację aktywowaną przez Abn-CBD.
Mniej badań skupiło się na badaniu wpływu CBD na naczynia krwionośne. Jarai i współpracownicy [25] nie stwierdzili wpływu perfuzji 10 µM CBD na tonus naczyniowy w zwężonym łóżku naczyniowym krezkowym szczura za pomocą fenylefryny. Niemniej jednak w segmencie tętniczym pobranym z łóżka naczyniowego krezkowego szczura, który został zamocowany na myografie Mulvany-Halpern i zwężony fenylefryną, CBD powoduje koncentracjopochodne, zależne od naczyń, niemal maksymalne rozszerzenie naczyń [28]. Niestety w tym badaniu nie zbadano mechanizmów leżących u podstaw tego efektu rozszerzającego naczynia CBD w tętnicach krezkowych szczura.
W tętnicach krezkowych u ludzi, bardzo niedawno pokazaliśmy, że CBD powoduje rozszerzenie naczyń segmentów tętniczych wcześniej zwężonych przez U46619 i endotelinę-1 (Stanley & O’Sullivan, 2012, w trakcie recenzji). W tętnicach krezkowych u ludzi, CBD-indukowane rozszerzenie naczyń ma pEC50 w zakresie środkowomikromolarnym, co jest podobne do tego obserwowanego w tętnicach krezkowych szczura. Jednakże CBD-indukowane rozszerzenie naczyń w tętnicach ludzkich osiąga maksymalną odpowiedź na poziomie około 40% redukcji przedimponowanego tonu. Udowodniliśmy również, że CBD-indukowane rozszerzenie naczyń w tętnicach krezkowych u ludzi jest zależne od śródbłonka, wiąże się z aktywacją receptora CB1 i aktywacją kanałów TRPV, uwolnieniem tlenku azotu oraz hiperpolaryzacją potasową (Rysunek 1) (Stanley & O’Sullivan, 2012, w trakcie recenzji).
Figura 1.
Bezpośrednie efekty naczyniowe CBD mierzone w izolowanych tętnicach. TRPV, przemijający receptor potencjałowy waniloidowy; NO, tlenek azotu; CB1, receptor kannabinoidowy 1; PPARγ, gamma receptor aktywowany przez proliferator peroksysomów; SOD, dysmutaza ponadtlenkowa.
Warto zauważyć, że niedawno Ruiz-Valdepenas i inni [34] pokazali, że CBD hamuje lipopolisacharydowy (LPS)-indukowany rozszerzający naczynia efekt arteriol i żylaków. Sugerowano, że LPS może powodować hipotensję poprzez aktywację nowego, jeszcze nie zidentyfikowanego receptora kannabinoidowego, który mógłby być zahamowany przez SR141716A, ale nie przez AM251 [35]. Ponieważ CBD jest sugerowany jako antagonista tego receptora [25], może to wyjaśniać, dlaczego CBD hamuje LPS-indukowany rozszerzający naczynia efekt.
Agonisty receptora PPARγ wykazano, że mają pozytywne efekty na układ sercowo-naczyniowy, obejmujące zwiększoną dostępność tlenku azotu, obniżenie ciśnienia krwi oraz złagodzenie miażdżycy [36, 37]. Niektóre korzystne efekty ligandów PPARγ wynikają z działań przeciwzapalnych, takich jak hamowanie cytokin prozapalnych, zwiększanie cytokin przeciwzapalnych i hamowanie ekspresji indukowalnej syntazy tlenku azotu (iNOS) (dla przeglądu, zobacz [38]). Narastające dowody wskazują, że kannabinoidy są zdolne do wiązania się, aktywowania i wywoływania odpowiedzi mediowanej przez PPAR [39]. Wykazaliśmy, że główny aktywny składnik konopi, THC, aktywuje PPARγ, a THC powoduje czasowo zależne, śródbłonowo zależne, PPARγ-zależne rozszerzenie tętniczej aorty szczura [40, 41]. Następnie przetestowaliśmy, czy CBD może również aktywować PPARγ i czy to może pośredniczyć w niektórych efektach farmakologicznych kannabidiolu. W tych eksperymentach pokazaliśmy, że CBD jest słabym/częściowym agonistą receptora PPARγ, który zwiększa aktywność transkrypcyjną PPARγ w komórkach HEK293 z nadmiernym wyrażeniem PPARγ, a CBD wiąże się z domeną wiążącą ligand PPARγ z IC50≍ 5 µm[5]. Podobnie jak THC, CBD (w stężeniach powyżej 100 nM) wykazał również czasowo zależne rozszerzenie tętniczej aorty szczura. To czasowo zależne rozszerzenie zostało zahamowane przy użyciu antagonisty PPARγ GW9662 lub inhibitory dysmutazy ponadtlenkowej (SOD) dietylditiokarbaminianu (DETCA). Zwiększona aktywność SOD sprzyja rozszerzeniu naczyń poprzez redukcję reaktywnych form tlenu, a nasze dane są zgodne z innymi badaniami, które pokazują, że ligandy PPARγ powodują indukcję Cu/Zn-SOD [42]. Należy jednak zauważyć, że ostatnie prace sugerują, że stosowanie leków z grupy tiazolidynodonów (TZD) może prowadzić do obniżenia funkcji układu sercowo-naczyniowego i może prowokować incydenty takie jak ostre zawały mięśnia sercowego, niewydolność serca i udar [43–45]. Skutki uboczne związane z ligandami PPARγ obejmują przyrost masy ciała, obrzęk i zwiększenie poziomu lipidów w osoczu [46]. Jednakże słabi/częściowi agonisty receptora PPARγ mogą być pozbawieni tych szkodliwych skutków ubocznych [46]. CBD może okazać się mieć potencjał terapeutyczny jako agonista o niskim powinowactwie do PPARγ.
Hemodynamiczne efekty CBD.
Do tej pory przeprowadzono niewiele badań dotyczących reakcji hemodynamicznych na CBD. Jedno badanie wykazało, że u szczurów znieczulonych pentobarbitaliem CBD (w dawce 50 µg/kg i.v., ale nie 10 µg/kg) powoduje znaczący, ale przejściowy spadek ciśnienia krwi o 16 mmHg, nie wpływając jednocześnie na częstość akcji serca [47]. Jednakże inne badania nie donoszą o żadnych ostrej reakcji na leczenie CBD na częstość akcji serca ani ciśnienie krwi w badaniach na zwierzętach [48, 49]. W niedawnym przeglądzie, Bergamaschi i inni [13] doszli do wniosku, że leczenie CBD u ludzi nie prowadziło do zmian ciśnienia krwi ani częstości akcji serca. Stąd większość dowodów sugeruje brak wpływu CBD na hemodynamikę. Niemniej jednak, podobnie jak obserwowano to w przypadku innych związków kannabinoidowych, potencjalne hipotensyjne działania CBD mogą ujawnić się w modelach podwyższonego ciśnienia krwi. Dodatkowo, jakiekolwiek zmiany hemodynamiczne mogą być szybkie [47] i dlatego nie zaobserwowane w badaniach nad przewlekłym leczeniem.
CBD jest znane z działania przeciwlękowego. Leczenie CBD redukuje lęk związany z publicznym przemawianiem lub bodźcami wywołującymi strach u ludzi [10]. Wiele badań wykazało również, że CBD redukuje odpowiedź sercowo-naczyniową na sytuacje stresowe lub sytuacje związane z lękiem. Resstel i jego koledzy wykazali na szczurach Wistar, że pojedyncza dawka CBD (10 lub 20 mg/kg i.p.) zmniejszyła odpowiedź częstości akcji serca i ciśnienia krwi na warunkowe lęki [49] lub na ostre ograniczenie ruchu [48]. Efekt hamujący CBD na odpowiedź sercowo-naczyniową na stres wydaje się być zahamowany przez WAY100635, antagonistę receptora 5HT1A. Ten efekt zdaje się być pośredniczony w mózgu, ponieważ ten sam efekt CBD na odpowiedź sercowo-naczyniową mógł być imitowany, gdy CBD było podawane do łóżka jądra pasma końcowego (struktury limbicznej) [50]. Potencjalna zdolność leczenia CBD u ludzi do redukcji odpowiedzi sercowo-naczyniowej (a także behawioralnej) na stres może mieć istotne skutki dla rozwoju miażdżycy i nadciśnienia, które są znane z przyspieszenia przez stres [51, 52].
Kardioprotekcyjne efekty CBD.
Kilka badań wykazało, że CBD jest korzystny w zapobieganiu uszkodzeniom niedokrwiennym-powodowanym wątroby [53, 54] i mózgu [55]. W 2007 roku Durst i jego koledzy po raz pierwszy pokazali, że leczenie CBD in vivo (5 mg/kg i.p. przed niedotlenieniem, a następnie przez 7 dni po nim) znacząco zmniejszyło wielkość obszaru zawału serca, gdzie związano lewą przednią zstępującą (LAD) tętnicę wieńcową, co wiązało się z redukcją infiltracji leukocytów i stężenia cyrkulującego interleukiny (IL)-6. Ponadto pokazali, że ten efekt kardioprotekcyjny CBD nie mógł być odtworzony in vitro, sugerując, że efekty kardioprotekcyjne CBD wynikają z ogólnego efektu immunomodulacyjnego, a nie bezpośredniego wpływu na serce [56]. Walsh i inni [47] później pokazali, że pojedyncza dawka CBD (50 µg/kg i.v.) podana 10 minut przed niedokrwieniem lub 10 minut przed reperfuzją znacząco zmniejszyła wielkość obszaru zawału po związaniu tętnicy wieńcowej LAD. Wiązało się to również z redukcją komorowych skurczów przedwczesnych, sugerując dodatkową rolę przeciarytmiczną CBD. Rajesh i inni [57] wykazali, że 11-tygodniowe leczenie CBD in vivo (20 mg/kg i.p.) znacząco zmniejszyło dysfunkcję serca u myszy z cukrzycą, związane z zmniejszoną zapalnością mięśnia sercowego, stresem oksydacyjnym, stresem azotowym i bliznowaceniem, pośredniczącym w zmniejszonej aktywacji czynnika jąderkowego κB (NFκB), zmniejszonej aktywacji kinazy białkowej aktywowanej przez mitogen (MAPK) i zmniejszonej ekspresji cząsteczek adhezyjnych i czynnika martwicy nowotworów (TNF). Inne badania wykazały, że efekty przeciwzapalne CBD poprzez NFκB nie są pośredniczone przez aktywację receptorów CB1, CB2 ani Abn-CBD [58].
Wszystkie te dane sugerują, że leczenie CBD in vivo wykazuje istotne efekty kardioprotekcyjne, które mogą wynikać z bezpośredniego oddziaływania na serce lub za pośrednictwem ogólnego mechanizmu przeciwpowstępowego i przeciwutleniającego (patrz Tabela 1).
Tabela 1
Obecna baza dowodów popierająca terapeutyczną rolę CBD w zaburzeniach sercowo-naczyniowych.
Naczynioprotekcyjne efekty CBD.
Narasta zbiór dowodów wskazujących, że podawanie CBD może złagodzić negatywne skutki warunków związanych z dysfunkcją śródbłonka. Wysokie stężenia glukozy związane z cukrzycą zostały zgłoszone jako czynnik przyczyniający się do dysfunkcji śródbłonka. Wysoka glukoza sprzyja hamowaniu/rozłączaniu tlenku azotu śródbłonkowego, zwiększonej produkcji nadtlenku azotu, zwiększonym działaniom zwężających prostanoizoidów, zmniejszonym działaniom rozszerzających prostanoizoidów oraz zwiększonym reaktywnym formom tlenu [59]. Oprócz tych zmian, wysokie stężenia glukozy są również zgłaszane jako czynnik zwiększający adhezję leukocytów i migrację monocytów przez śródbłonek [60], co miało miejsce poprzez aktywność NFκB [61].
W komórkach śródbłonka ludzkich tętnic wieńcowych, długotrwałe wystawienie na wysokie stężenia glukozy spowodowało zwiększone poziomy cząsteczek adhezyjnych (ICAM-1 i VCAM-1), zakłócenie bariery śródbłonka, produkcję nadtlenku azotu mitochondrialnego, ekspresję iNOS oraz NFκB [62]. Wszystkie te efekty zostały zredukowane, gdy komórki były współinkubowane z CBD w porównaniu z samą wysoką glukozą. CBD również zmniejszyło adhezję monocytów i migrację tranśendotelialną, które są kluczowe w postępowaniu miażdżycy. Ani receptory CB1, ani CB2 nie były odpowiedzialne za pośredniczenie w efektach CBD [62]. Korzystając z modelu in vivo retinopatii cukrzycowej, El-Remessy i inni [63] podobnie stwierdzili, że leczenie CBD (10 mg/kg, co 2 dni, i.p.) zapobiegało nadmiernemu przepuszczaniu naczyń krwionośnych na barierze krew–siatkówka (BRB) i chroniło siatkówkę przed uszkodzeniem oksydacyjnym, stanem zapalnym oraz wzrostem cząsteczek adhezyjnych. Stąd ochrona naczyń krwionośnych przez CBD w modelu cukrzycy może prowadzić do zmniejszenia powikłań, takich jak retinopatia, choć może być to również wynikiem neuroprotekcyjnych efektów CBD.
Zakaźne zapalenie mózgu, modelowane poprzez parenteralne podanie LPS myszom, indukuje głęboką rozszerzalność arterioli, prowadzącą do nadmiernego ukrwienia mózgu i zakłócenia barierki krew-mózg (BBB) [34]. Podawanie CBD (3 mg/kg i.v.) jednocześnie z LPS utrzymało integralność BBB, zahamowało LPS-indukowaną rozszerzalność arterioli i żyłek, marginesację leukocytów oraz stłumiło nadmierne wydzielanie tlenku azotu. Choć bezpośrednio nie mierzono przepływu krwi w mózgu (CBF), wyniki obserwowane z różnych parametrów skłoniły autorów do sugerowania, że CBD złagodziło spadek CBF wywołany przez LPS [34].
Przeprowadziliśmy pewne wstępne badania dotyczące zdolności CBD do modulowania odpowiedzi naczyniorozszerzającej. Wykorzystując szczura Zucker, będącego modelem cukrzycy typu 2, w którym wiadomo, że jest upośledzona śródbłonkowa naczyniorozszerzalność, wykazaliśmy, że inkubacja aorty przez 2 godziny z CBD (10 µm) istotnie zwiększała odpowiedź naczyniorozszerzającą na acetylocholinę, endotelium-zależny naczyniorozszerzacz [64]. Podobnie wykazaliśmy, że inkubacja z CBD zwiększa odpowiedź naczyniorozszerzającą na acetylocholinę u spontanicznie nadciśnieniowego szczura (O’Sullivan, dane niepublikowane).
Podsumowując, te badania pokazują, że in vitro i in vivo, stosując hodowlę komórkową, izolowane tkanki i modele zwierzęce, CBD zostało wykazane, że zmniejsza negatywne skutki wysokich stężeń glukozy, cukrzycy i stanów zapalnych na naczynia krwionośne i na nadmierną przepuszczalność naczyń. Dotychczasowe badania nie jednoznacznie określają receptory, na które CBD oddziałuje w niektórych z tych badaniach, ale wspólnym motywem jest redukcja markerów zapalnych (patrz Tabela 1).
CBD w modelach udaru
Podawanie endogennych, syntetycznych lub fitokannabinoidów zostało wykazane jako środek neuroprotekcyjny w różnorodnych modelach chorób in vivo i in vitro, w tym w udarze [65]. Potencjał neuroprotekcyjny CBD w udarze niedokrwiennym został po raz pierwszy zbadany przez Hampson i współpracowników [66], gdzie szczury poddano zamknięciu tętnicy środkowej mózgu (MCAO), a CBD podane w chwili wystąpienia incydentu (5 mg kg−1, i.v.) oraz 12 godzin po zabiegu (20 mg kg−1, i.p.) zmniejszyło rozmiar obszaru niedokrwionego i upośledzenie neurologiczne o 50–60%. Podobnie, podanie CBD po udarze niedokrwiennym (1.25 do 20 mg kg−1, i.p.) chroniło przed utratą fal elektroencefalograficznych, hiperlokomocją i uszkodzeniem neuronalnym u myszy z MCAO [67]. Ostatnio wykazano, że CBD (3 mg kg−1, i.p.) redukuje objętość obszaru niedokrwionego po MCAO, niezależnie od receptora CB1 lub TRPV1, ale jest wrażliwe na antagonistę receptora 5HT1A WAY100135 (10 mg kg−1, i.p.) [68, 69]. Ponadto CBD (3 mg kg−1, i.p.) zapewniło neuroprotekcję nawet w przypadku podania do 2 godzin po reperfuzji, bez wytworzenia tolerancji [70, 71].
Przepływ krwi w mózgu jest zmniejszony lub całkowicie zablokowany w niektórych obszarach mózgu podczas udaru niedokrwiennego, dlatego przywrócenie odpowiedniego przepływu krwi jest kluczowe. CBD okazało się skuteczne w zwiększaniu przepływu krwi, mierzonego za pomocą laserowej dupleksy metody Dopplera, po MCAO i po reperfuzji (3 mg kg−1, i.p.) [69–71]. Zwiększony przepływ krwi indukowany przez CBD (3 mg kg−1, i.p.) był częściowo zmniejszony przez blokadę receptora 5HT1A, sugerując, że CBD może wywierać te korzystne efekty, przynajmniej częściowo, poprzez receptor serotoninowy 5HT1A [69]. Ekspozycja noworodków na hipoksję i niedotlenienie, przeprowadzona przez Alvareza i współpracowników [72], również wykazała zdolność CBD (0.1 mg kg−1, i.v., po urazie) do zapewnienia neuroprotekcji, obejmującej zachowanie krążenia mózgowego.
Jak wcześniej omawiano, podawanie CBD (3 mg kg−1, i.v.) jednocześnie z LPS utrzymuje integralność BBB [34]. Chociaż przepływ krwi mózgowej nie był bezpośrednio mierzony, wyniki obserwowane na podstawie różnych parametrów skłoniły autorów do sugestii, że CBD złagodziło spadek przepływu krwi mózgowej indukowany przez LPS [34]. Uszkodzenie BBB jest istotnym elementem w patofizjologii udaru niedokrwiennego [73]. Dlatego utrzymanie przez CBD tej bariery, jak wykazano w innych modelach chorób, może stanowić kolejny mechanizm ochrony CBD w udarze niedokrwiennym. Agonizm PPARγ może reprezentować kolejny mechanizm działania korzystnych efektów CBD w udarze. Wiele grup wykazało, że syntetyczne agonisty PPARγ, tiazolidynodiony (TZD), klasa leków stosowana w poprawie wrażliwości na insulinę, zmniejszają obszar niedokrwienia i poprawiają zdolność do funkcji po udarze u szczurów [74–78]. Poprawa ta jest związana z redukcją stanu zapalnego, który jest prawdopodobnym mechanizmem poprawy, a co najważniejsze, poprawa jest widoczna zarówno w przypadku podawania TZD przed, jak i po MCAO [75, 77]. Ostatnio pokazano, że leczenie CBD in vivo wykazuje efekty neuroprotekcyjne w modelu choroby Alzheimera, które były hamowane przez antagonistę PPARγ [6]. Podobnie pokazaliśmy w modelu kultury komórkowej BBB, że CBD przywraca zwiększoną przepuszczalność indukowaną przez niedotlenienie glukozy, co można było zahamować za pomocą antagonisty PPARγ (Hind & O’Sullivan, niepublikowane obserwacje).
Podsumowując, CBD zapewnia neuroprotekcję w modelach zwierzęcych i in vitro udaru. Oprócz bezpośrednich efektów neuroprotekcyjnych CBD, działanie to jest pośredniczone zdolnością CBD do zwiększenia przepływu krwi mózgowej i zmniejszenia nadmiernego przepuszczania naczyń krwionośnych w mózgu (patrz Tabela 1).
Haematologiczne efekty CBD
Dotychczasowe badania nad wpływem CBD na układ krwiotwórczy skoncentrowały się głównie na komórkach białych, migracji komórek białych i agregacji płytek krwi. CBD wykazuje wpływ na przeżycie i śmierć białych krwinek, migrację komórek białych oraz agregację płytek krwi.
Wpływ CBD na komórki białe krwi: CBD wpływa na przeżycie i śmierć komórek białych krwi, migrację tych komórek oraz agregację płytek krwi. Badania wykazują, że CBD wpływa na białe krwinki, regulując ich przeżycie i śmierć, migrację oraz zdolność do agregacji. Skutki te są zazwyczaj korzystne, prowadząc do zmniejszenia stanów zapalnych.
Wpływ CBD na migrację komórek białych: CBD ma wpływ na migrację komórek białych, co odgrywa istotną rolę w reakcjach zapalnych. Wpływ ten zazwyczaj zmniejsza zdolność komórek białych do przenikania do obszarów zapalnych, co może być korzystne w kontekście redukcji stanów zapalnych.
Wpływ CBD na agregację płytek krwi: CBD ma również wpływ na agregację płytek krwi, co może wpływać na proces krzepnięcia. Wpływ ten może być korzystny w kontekście redukcji ryzyka powstawania skrzepów.
Podsumowując, badania nad wpływem CBD na układ krwiotwórczy sugerują, że kannabidiol może mieć korzystny wpływ na komórki białe krwi, migrację komórek białych i agregację płytek krwi, co może przyczyniać się do zmniejszenia stanów zapalnych oraz ryzyka powstawania skrzepów. Jednak potrzebne są dalsze badania, aby lepiej zrozumieć mechanizmy działania i potwierdzić te efekty, zwłaszcza w kontekście wpływu CBD na ludzki organizm.
Wnioski
Podsumowując, niniejsza recenzja przedstawiła dowody na pozytywne efekty kannabidiolu (CBD) w układzie sercowo-naczyniowym, które zostały podsumowane w Tabeli 1. W izolowanych tętnicach bezpośrednie zastosowanie CBD powoduje zarówno ostry, jak i czasowy efekt rozszerzenia naczyń krwionośnych przedwężonych, a także wzmacnia rozszerzalność naczyń zależną od śródbłonka w modelach dysfunkcji śródbłonka. W warunkach in vivo leczenie CBD nie wydaje się mieć wpływu na spoczynkowe ciśnienie krwi ani na częstość akcji serca, ale zmniejsza reakcję układu sercowo-naczyniowego na różne rodzaje stresu. CBD odgrywa ochronną rolę w redukowaniu efektów niedokrwienia i reperfuzji serca, a także w zmniejszaniu dysfunkcji serca związanego z cukrzycą. Podobnie CBD pełni ochronną rolę w redukowaniu szkód niedokrwiennych w modelach udaru mózgu, częściowo poprzez utrzymanie przepływu krwi w mózgu. W modelach zmienionej przepuszczalności naczyń, CBD zmniejsza nadmierną przepuszczalność barier w cukrzycy i po podaniu LPS zmniejsza przepuszczalność barier w mózgu. Podsumowując, te dane sugerują, że układ sercowo-naczyniowy stanowi rzeczywisty cel terapeutyczny dla CBD. Niemniej jednak miejsca działania CBD w większości tych reakcji pozostają do ustalenia. Czy te odpowiedzi na CBD przekładają się również na ludzki układ sercowo-naczyniowy, również pozostaje do ustalenia.
Układ endokannabinoidowy (eCB) został dokładnie zbadany w celu zidentyfikowania struktur molekularnych obecnych w Cannabis sativa. eCB składa się z receptorów kannabinoidowych, endogennych ligandów i związanej z nimi aparatury enzymatycznej odpowiedzialnej za utrzymanie homeostazy energetycznej i procesów poznawczych. Kilka efektów fizjologicznych kannabinoidów jest wywieranych poprzez interakcje z różnymi receptorami, takimi jak receptory CB1 i CB2, receptory waniloidowe, a także niedawno odkryte receptory sprzężone z białkiem G (GPR55, GPR3, GPR6, GPR12 i GPR19). Anandamid (AEA) i 2-arachidonoylglicerol (2-AG), dwa małe lipidy pochodzące od kwasu arachidonowego, wykazały wysokie powinowactwo do receptorów CB1 i CB2. eCB odgrywa kluczową rolę w przewlekłym bólu i zaburzeniach nastroju, dlatego został szeroko zbadany ze względu na swoje szerokie potencjalne zastosowanie terapeutyczne i jako obiecujący cel dla rozwoju nowych leków. Fytokannabinoidy i kannabinoidy syntetyczne wykazały zróżnicowane powinowactwo do eCB i są istotne w leczeniu wielu chorób neurologicznych. Niniejsza recenzja opisuje składniki eCB i omawia, w jaki sposób fytokannabinoidy i inne związki egzogenne mogą regulować równowagę eCB. Ponadto przedstawiamy hipo- lub hiperfunkcjonalność eCB w organizmie oraz związki eCB z przewlekłym bólem i zaburzeniami nastroju, nawet przy zastosowaniu praktyk zdrowotnych zintegrowanych i uzupełniających (ICHP) harmonizujących eCB.
Wprowadzenie Cannabis sativa jest używana do celów rekreacyjnych [1,2], terapeutycznych [3,4] oraz innych od tysięcy lat [5]. Roślina zawiera ponad 120 terpenów zwanych fitokannabinoidami, w tym jednego z głównych i najbardziej rozpoznawalnych przedstawicieli, Δ9-tetrahydrokannabinolu (THC). Strukturę molekularną Δ9-THC zidentyfikowano po raz pierwszy w 1964 roku, co prowadziło do przypuszczenia istnienia receptora kannabinoidowego i zwiększyło odkrywanie układu endokannabinoidowego (eCB), który głównie jest systemem międzykomórkowym odpowiedzialnym za homeostazę energetyczną i regulację spożycia pokarmu, metabolizmu i wydatków energetycznych, utrzymując stałą masę ciała [6]. Oprócz apetytu, eCB może przyczyniać się do procesów poznawczych związanych z pamięcią, nastrójem i bólem [7]. eCB zyskał na znaczeniu podczas pandemii COVID-19, nie tylko ze względu na hamowanie replikacji SARS-CoV-2, ale także w różnych badaniach obejmujących jego zastosowanie w leczeniu przewlekłego bólu i zaburzeń nastroju [8,9,10,11,12,13]. eCB to aktywny system, który pobudza złożoną sieć sygnalizacji komórkowej. Obejmuje on kombinację receptorów kannabinoidowych, endogennych kannabinoidów (endokannabinoidów) i enzymów odpowiedzialnych za syntezę i degradację endokannabinoidów. Pierwsze badania rozpoczęły się od identyfikacji receptorów o nazwach receptor kannabinoidowy typu 1 i 2, czyli CB1R i CB2R [14,15,16,17]. Ponadto dokonano odkrycia endogennych ligandów, co wzbogaciło naszą wiedzę o nowe związki, takie jak N-arachidonoyletanoloamid, pierwszą cząsteczkę endokannabinoidu odkrytą, nazwaną „anandamidem”, słowem sanskryckim oznaczającym „błogość” lub ekstremalne szczęście [18], a następnie zidentyfikowano 2-arachidonoyloglicerol (2-AG), które wraz z enzymami odpowiedzialnymi za syntezę i degradację tych związków stanowią to, co dzisiaj nazywamy układem endokannabinoidowym [19]. W październiku 2022 r. wyszukiwanie w bazie PubMed artykułów naukowych opublikowanych we wszystkich dostępnych okresach, zawierających słowo „Endocannabinoid”, wykazało 12 272 wyniki, zwiększające się zainteresowanie badaniami i publikacjami od roku 2000. Te liczby ilustrują szybko rosnące wsparcie finansowe w ostatnich latach oraz zainteresowanie naukowe zrozumieniem mechanizmów molekularnych w różnych kontekstach zastosowań klinicznych. Niniejsza recenzja skupia się na ostatnich postępach w zrozumieniu składników eCB i omawia role fitokannabinoidów, innych związków egzogennych, leczenie bólu i zaburzeń nastroju przy użyciu eCB, a także praktyki zdrowotne zintegrowane i uzupełniające.
Receptory kannabinoidowe Receptory kannabinoidowe CB1/CB2 są głównie odróżniane przez sekwencję aminokwasów w łańcuchu polipeptydowym i przez ich rozmieszczenie w różnych tkankach [20,21,22,23] (Rysunek 1). Badania farmakologiczne sugerują, że cząsteczki kannabinoidowe mogą oddziaływać na receptory inne niż klasyczne receptory CB1 i CB2, takie jak receptory waniloidowe TRPV1, TRPV2, TRPV3, TRPV4, TRPA1, TRPM8 oraz receptory metabotropowe, takie jak GPR55, GPR3, GPR6, GPR12 i GPR19, między innymi receptory, a także enzymy i białka [24,25,26]. Ostatnio eCB został rozwinięty, a badacze nazwali go endokannabinoidom (eCBome), znaczącym odniesieniem obejmującym wszystkie składniki oraz białka, enzymy i lipidy, które bezpośrednio lub pośrednio uczestniczą w modulacji systemu kannabinoidowego i istotnie wpływają na zdrowie [27].
Rysunek 1. Receptory kannabinoidowe CB1 i CB2 oraz ich rozmieszczenie w ludzkim ciele.
Endogenny system sygnalizacji lipidowej może być głęboko zaangażowany w wiele stanów fizjologicznych i zaburzeń patologicznych, co może stanowić perspektywę dla przyszłego leczenia różnych chorób [28,29,30,31]. Fitokannabinoidy, takie jak kannabidiol (CBD), wykazują szerokie zastosowanie terapeutyczne, prawdopodobnie ze względu na zdolność do oddziaływania na liczne receptory. eCBome odgrywa rolę w osi mikrobiota-jelito-mózg, która stała się ważnym uczestnikiem kontroli funkcji afektywnych i poznawczych oraz ich zmian patologicznych. Jednak molekularne i biochemiczne podstawy interakcji oraz biologiczne zależności nowych podtypów receptorów z ligandami kannabinoidowymi nie zostały w pełni wyjaśnione; dlatego konieczne są dalsze badania [32,33,34]. Receptor kannabinoidowy typu 1 (CB1R) jest kodowany przez gen CNR1 [35,36] i został sklonowany u szczurów przez Matsudę i innych w 1990 roku [14]. Lata później CB1R został również sklonowany w ludzkich tkankach [37,38] oraz u myszy [39], wykazując 97–99% tożsamości sekwencji aminokwasów między tymi gatunkami [40]. Po sklonowaniu receptora możliwe było zaprojektowanie cząsteczek ligandów, które pasują do tych receptorów, stosując logikę modelu klucz-zamek [14,41,42,43]. Radioaktywny środek śledzący syntezowany przez Pfizer („CP55, 940”) umożliwił badaczom zmapowanie lokalizacji receptorów kannabinoidowych w mózgu. Receptory te zostały zidentyfikowane w ośrodkowym układzie nerwowym (OUN) i w wysokich stężeniach w regionach odpowiedzialnych za procesy psychiczne i fizjologiczne, takie jak hipokamp (pamięć), kora mózgowa (poznawczość), móżdżek (koordynacja ruchowa), jądra podstawy (ruch), podwzgórze (apetyt) i migdał (emocje) [35,44]. Receptory kannabinoidowe występują w mniejszej liczbie w jądrze mostowym, regionie kontrolującym oddychanie i akcję serca, co może wyjaśniać brak doniesień o zgonach z powodu przedawkowania Cannabis, niezależnie od wieku, celu klinicznego lub drogi podania [45,46]. CB1R jest również wyrażany w komórkach glejowych, oligodendrocytach i mikrogleju, które uczestniczą w przewodnictwie synaptycznym [46,48,49]. Dotychczasowe badania sugerują, że CB1R jest wysoce wyrażany w zakończeniach presynaptycznych i moduluje retrogradne sygnały endokannabinoidowe [50]. Jednak istnienie CB1R na miejscach postsynaptycznych nie zostało wykluczone, na przykład w badaniach funkcjonalnych wykazujących autoinhibicję neuronów korowych przez endokannabinoidy [51]. Badania dotyczące mapowania mózgu szczura sugerują, że preferowanym miejscem CB1R są aksony i zakończenia nerwowe, a jego działania związane są z regulacją uwalniania neurotransmiterów, takich jak noradrenalina, dopamina, acetylocholina, glutaminian, 5-hydroksytryptamina, kwas γ-aminomasłowy (GABA) i d-asparaginian [1,52,53]. CB1R jest obficie wyrażany także w obwodowym układzie nerwowym, jak i w innych obszarach ciała [54,55]. W układzie nerwowym obwodowym CB1R jest wysoce wyrażany w zakończeniach nerwów współczulnych. Ponadto CB1R występuje w zwojach trójdzielnych, zwojach korzeni tylnych i zakończeniach nerwów skórnych pierwszorzędowych neuronów czuciowych, gdzie reguluje nocicepcję włókien nerwowych aferentnych. W przewodzie pokarmowym (GIT) CB1R jest wyrażany zarówno w układzie nerwowym jelit, jak i w komórkach nie-nerwowych, takich jak błona śluzowa jelita, w tym komórki enteroendokrynne, komórki immunologiczne i enterocyty. CB1R reguluje ruchliwość GIT [56], wydzielanie kwasów żołądkowych [57], płynów [58], neurotransmiterów [59] i hormonów [60], a także przepuszczalność nabłonka jelitowego [61]. CB1R obecny w OUN odgrywa rolę w regulacji apetytu w podwzgórzu i reguluje bilans energetyczny oraz spożycie pokarmu w GIT. Co ciekawe, wątrobowe CB1R mogą również brać udział w regulacji bilansu energetycznego i metabolizmu [46,62,63]. Normalnie ekspresja CB1R w wątrobie jest bardzo niska; jednak w warunkach patologicznych ekspresja CB1R w różnych rodzajach komórek wątroby jest znacznie zwiększona, gdzie CB1R aktywnie przyczynia się do insulinooporności wątrobowej, włóknienia i lipogenezy. Podobnie CB1R jest regulowany w układzie sercowo-naczyniowym w warunkach patologicznych, co sprzyja postępowi choroby i dysfunkcji serca [55,64,65,66]. Aktywacja CB1R w kardiomiocytach, komórkach śródbłonka naczyń krwionośnych i komórkach mięśni gładkich prowadzi do stresu oksydacyjnego, stanu zapalnego i włóknienia. Oprócz wspomnianych tkanek, ekspresję CB1R stwierdzono także w tkance tłuszczowej, mięśniach szkieletowych, kościach, skórze, oczach, układzie rozrodczym oraz różnych typach komórek nowotworowych. W mięśniach szkieletowych i miokardium receptory CB1R są głównie zlokalizowane w mitochondriach (mtCB1R). Aktywacja receptorów mtCB1 może uczestniczyć w regulacji mitochondrialnej aktywności oksydacyjnej, prawdopodobnie poprzez istotne enzymy zaangażowane w metabolizm kwasu pirogronowego, głównego substratu dla aktywności cyklu trójkarboksylowego [23,67,68,69]. Receptor kannabinoidowy typu 2 (CB2R) został sklonowany w 1993 roku z komórek promielocytarnego białaczka ludzkiej linii HL-60 [15] i dalej zidentyfikowany u myszy, szczurów, ryb karpiowatych i psów [70,71,72,73]. Ma on sekwencję aminokwasów o około 44% homologii z resztami aminokwasów CB1R. CB2R znajduje się głównie w komórkach układu immunologicznego, gdzie poziomy jego ekspresji są wyższe niż CB1R [24,46,74]. CB2R reguluje komórki immunologiczne i przyczynia się do efektów analgetycznych i/lub przeciwnociceptywnych kannabinoidów. CB2R został zidentyfikowany także w OUN. Jednak niektóre badania wykazały jego obecność na powierzchni mikrogleju i neuronów zlokalizowanych w móżdżku, pniu mózgu, wzgórzu, jądrze prążkowania, korze, migdałach i hipokampie [46,75]. Zarówno CB1R, jak i CB2R należą do dużej rodziny receptorów sprzężonych z białkami G (GPCR). Są to białka błonowe zbudowane z aminokwasowego domeny ekstracellularnego, siedmiu konserwatywnych helis o charakterystycznej sekwencji 20–27 reszt aminokwasowych o wysokiej hydrofobowości, trzech pętli ekstracellularnych i trzech pętli intracellularnych oraz domeny karboksylowej końca wewnątrzkomórkowego [40,76] (Rysunek 2).
Rysunek 2. Ilustracja receptora należącego do dużej rodziny receptorów sprzężonych z białkami G (GPCR).
Aktywacja obu receptorów kannabinoidowych prowadzi do hamowania cyklazy adenylanowej w różnych typach komórek poprzez sprzężenie z białkiem Gi/o. To prowadzi do obniżenia poziomów 3′,5′-monofosforanu adenozyny (cAMP) i aktywności kinazy białkowej A (PKA), co może być związane z nadwrażliwością neuronów nociceptywnych oraz białkami, które mogą być związane z zwiększonym stężeniem wapnia wewnątrzkomórkowego, trifosforanem mioinozytolu i diacyloglicerolami, które ostatecznie są zaangażowane w modulację uwalniania neurotransmiterów [77,78]. Stymulacja CB1R prowadzi do aktywacji szlaku sygnałowego kinazy białkowej aktywowanej przez mitogen (MAPK), w tym kinazy regulowanej sygnałowo przez czynniki zewnętrzne 1/2 (ERK1/2), kinazy N-terminalnej c-Jun (JNK) i p38, które są zaangażowane w regulację proliferacji komórkowej, kontrolę cyklu komórkowego i śmierć komórkową [46,79] (Rysunek 3).
Rysunek 3. Mechanizm sygnalizacji wewnątrzkomórkowej poprzez aktywację receptorów kannabinoidowych CB1 i CB2. AC—cyklaza adenylanowa, cAMP—cykliczny monofosforan adenozyny; ATP—trifosforan adenozyny; MAPK—kinaza białkowa aktywowana przez mitogen; PKA—kinaza białkowa A.
Endokannabinoidy: Synteza, uwalnianie i metabolizm Odkrycie receptorów kannabinoidowych wzbudziło zainteresowanie poszukiwaniem endogennych ligandów odpowiedzialnych za ich modulację. Ocena oczyszczonych frakcji mózgu świń doprowadziła do zidentyfikowania nowego związku, który wiąże się z receptorem CB1. Arachidonyletanolamid, pochodna kwasu arachidonowego w mózgu świń, został scharakteryzowany i nazwany anandamidem (AEA), słowem pochodzącym od sanskryckiego słowa „ananda”, oznaczającego skrajne szczęście [18,77,80,81,82].
Na podstawie rozwiązania struktury AEA zidentyfikowano inne endogenne cząsteczki lipidowe (Rysunek 4), zwane ogólnie N-acyletanolaminami (NAE), takie jak 2-arachidonoyloglicerol (2-AG), N-oleoyletanolamina (OEA), 2-arachidonylogliceryloeter (noladin, 2-AGE), wirodamina, N-arachidonoyletyloamina (NADA) i N-palmitoyletanolamina (PEA). AEA i 2-AG są najbardziej zbadanymi endokannabinoidami; jednak badania nad endokannabinoidami zostały kontynuowane, a odkryto dodatkowe receptory, wraz z ich mediatorami lipidowymi i ścieżkami sygnalizacyjnymi [81,83,84,85,86,87,88,89,90,91,92,93].
Rysunek 4. Struktury chemiczne głównych endokannabinoidów.
Endokannabinoidy, w przeciwieństwie do klasycznych neuroprzekaźników, są uważane za nietypowe przekaźniki ze względu na modulację informacji z zakończeń postsynaptycznych do zakończeń presynaptycznych, co jest znane jako mechanizm sygnalizacji zwrotnej. Endogenne ligandy są syntetyzowane na żądanie lub w wyniku aktywności zależnej od rozkładu błony fosfolipidowej i są natychmiast uwalniane po ich biosyntezie, aby działać jako czynniki pro-homeostatyczne poprzez oddziaływanie z określonymi receptorami [77,94,95].
Synteza i degradacja endogennych ligandów receptora kannabinoidowego obejmują różne reakcje enzymatyczne. Biosynteza AEA zachodzi poprzez jej uwalnianie z fosfolipidów błonowych i może przebiegać poprzez zależną od Ca2+ N-acylotransferazę (NAT) lub niezależną od Ca2+ ścieżkę N-acylotransferazę (iNAT). W wyniku tego N-arachidonoilo-fosfatydyloetanolaminy (NArPE) są tworzone, a przez działanie N-acylofosfatydyloetanolamino-specyficznej fosfolipazy D (NAPE-PLD), NArPE jest przekształcane w N-arachidonoiloetanolaminę (AEA) [55,96,97].
Inny endogenny ligand, 2-AG, jest tworzony za pomocą dwuetapowego mechanizmu. Początkowo 1,2-diacyloglicerol (DAG) jest syntetyzowany po rozkładzie fosfolipidu błonowego przez enzym fosfolipazy C (PLC). Następnie DAG jest estryfikowany przez enzym diacyloglicerolowa lipaza (DAGL), tworząc 2-AG [98,99].
Endogenne kannabinoidy stają się nieaktywne poprzez komórkowy mechanizm ponownego wychwytu za pomocą transporterów błonowych (EMT), a następnie ulegają intrakomórkowej degradacji pod wpływem enzymów hydrolitycznych. Anandamid głównie jest metabolizowany przez enzym hydrolazy amidu kwasu tłuszczowego (FAAH), a 2-AG jest substratem lipazy monoacyloglicerolowej (MAGL), która wytwarza kwas arachidonowy (AA) i glicerol [100,101] (Rysunek 5).
Rysunek 5. Szlaki metaboliczne zaangażowane w syntezę i degradację anandamidu (5) oraz 2-arachidonyloglicerolu (6). AA—kwas arachidonowy; 2-AG—2-arachidonyloglicerol; DAGL—lipaza diacyloglicerolowa; EMT—transportery błonowe; FAAH—hydrolaza amidu kwasu tłuszczowego; MAGL—lipaza monoacyloglicerolowa; NAPE-PLD—fosfolipaza D N-arachidonylofosfatydyloetanolaminy; NArPE—N-acylofosfatydyloetanolamina; NAT—N-acylotransferaza; PLC—fosfolipaza C.
Ponadto, AEA i 2-AG mogą być podatne na mechanizmy utleniania katalizowane przez cyklooksygenazy (COX), lipooksygenazy (LOX) i enzymy zaangażowane w utlenianie kwasu arachidonowego (AA), który ulega biotransformacji do prostaglandyn (PG), eikozanoidów oraz hydroksy-peroksy-anandamidu, między innymi związków pochodzących z tej reakcji metabolicznej [55,77].
Teoria niedoboru endokannabinoidów opiera się na koncepcji, że wiele zaburzeń mózgu jest związanych z niedoborem neuroprzekaźników, takich jak acetylocholina w chorobie Alzheimera (AD), dopamina w zespołach parkinsonowskich, a także serotonina i noradrenalina w depresji, podobny niedobór w poziomie endokannabinoidów może się manifestować w pewnych zaburzeniach, które wykazują przewidywalne cechy kliniczne jako skutki tego niedoboru [102,103,104].
W 2004 roku profesor dr Ethan Russo i jego współpracownicy zaproponowali zespół klinicznego niedoboru endokannabinoidów (CDS), sugerując, że zubożenie endokannabinoidów (hipofunkcjonalny eCB) może powodować wiele chorób, takich jak migrena, bardzo złożona choroba, która wiąże się z sygnalizacją między różnymi obszarami mózgu a różnymi neurochemicznymi przekaźnikami. Dokładna przyczyna migreny nie jest w pełni zrozumiała, chociaż predyspozycje genetyczne uważa się za główny czynnik przyczyniający się do jej powstawania i modyfikacji [102,104]. Możliwy związek między migreną a układem endokannabinoidowym został podkreślony przez kilka badań [105,106].
Fibromialgia również jest związana z niedoborami w układzie endokannabinoidowym i charakteryzuje się ostrym i przewlekłym rozległym bólem mięśniowo-szkieletowym w całym ciele. Ten ból często towarzyszy innym dolegliwościom, takim jak bezsenność, migrena, wahania nastroju, problemy z pamięcią, zespół jelita drażliwego (IBS) i przewlekłe zmęczenie. Charakterystyczne bolesne guzki, znane jako punkty spustowe, są zauważalne, zwłaszcza w ramionach i szyi. Podobnie jak w migrenie, fibromialgia wiąże się z hiperalgezją, czyli obniżonym progiem bólu związanym z hipofunkcją endokannabinoidów w rdzeniu kręgowym. Według Russo i innych, zatwierdzone leki na fibromialgię, duloksetyna, milnacipran (inhibitory serotoniny i adrenergii, odpowiednio) i pregabalina (przeciwpadaczkowe stosowane w leczeniu bólu neuropatycznego), wykazały niewielką skuteczność w porównaniu z kannabidoidami [106,107,108].
Zespół jelita drażliwego (IBS), znany również jako jelito kurczliwe, to zaburzenie czynnościowe charakteryzujące się bólem, skurczami, dyskomfortem i zmienionym ruchem jelit, głównie biegunką. Ruch, wydzielanie i stan zapalny w przewodzie pokarmowym są modulowane przez układ endokannabinoidowy, co uzasadnia stosowanie kannabinoidów w leczeniu IBS [109]. Badania wykazały, że zwiększone włókna czuciowe wyrażające receptory TRPV1 mogą przyczyniać się do nadwrażliwości trzewnej i bólu w IBS, co stanowi nowy cel terapeutyczny. Kannabidiol może być stosowany w interwencjach terapeutycznych ze względu na jego wpływ na receptory VR1 oraz zwiększanie sygnalizacji anandamidu. Jego analogi okazały się skutecznymi inhibitorami pobierania anandamidu przez komórki [110,111,112,113,114,115].
Zaburzenia neurodegeneracyjne mogą prowadzić do rozwoju choroby Parkinsona (PD) i AD. Zazwyczaj charakteryzują się one upośledzeniem funkcji poznawczych i innymi defektami neurologicznymi. Obecnie układ endokannabinoidowy jest badany jako cel terapeutyczny w PD i AD ze względu na nadmierne wyrażanie receptorów układu endokannabinoidowego, które wywierają neuroprotekcję przeciwko PD i zmniejszają neurozapalenie w AD. Stwierdzono zwiększone poziomy AEA w płynie mózgowo-rdzeniowym u nieleczonych pacjentów z PD, co sugeruje możliwy mechanizm kompensacyjny. Deficyty poznawcze u pacjentów z AD korelują z zaburzeniami wrażliwych obszarów mózgu, głównie w korze przedczołowej i hipokampie, które są bogate w CB1R. Δ9-THC i CBD wykazały neuroprotekcję w modelach zwierzęcych PD i AD; jednakże wywołały one toksyczne efekty u pacjentów, gdy były podawane bezpośrednio. Konieczne są badania w celu określenia skuteczności terapeutycznej ukierunkowanego na układ endokannabinoidowy leczenia chorób neurodegeneracyjnych [101,116,117,118].
W niektórych przypadkach układ endokannabinoidowy może być nadaktywny, co sprzyja zaburzeniom poznawczym obserwowanym w zespole łamliwego chromosomu X (FXS), zespole Downa i zespole Williamsa–Beurena (WBS). Oprócz przyczyn genetycznych tych zespołów wierzy się, że układ endokannabinoidowy jest nadaktywowany. W modelu zwierzęcym FXS, myszy z wyłączonym białkiem łamliwego chromosomu X (Fmr1) odtwarzają główne cechy choroby. Blokowanie CB1R i CB2R u samców myszy z wyłączonym Fmr1 normalizowało upośledzenie poznawcze i zachowanie przeciwlękowe, odpowiednio [119]. W preklinicznym modelu zespołu Downa segmentalnie trizomiczne myszy modelowe Ts65Dn wykazały, że ekspresja CB1R została wzmocniona, a jej funkcja zwiększona w synaptycznych zakończeniach w hipokampie. Redukcja ekspresji i blokada CB1R naprawiły deficyty pamięci u samców myszy Ts65Dn [120]. Aby ocenić model imitujący WBS, użyto myszy z tym samym usunięciem genetycznym, jak u pacjentów z WBS. Samce myszy wykazywały hiperaktywne zachowania społeczne, deficyty pamięci, powiększone serca i różnice w funkcji CB1R. Te myszy mutacyjne otrzymały JZL184, inhibitor MAGL, co poprawiło ich objawy społeczne i pamięciowe oraz funkcję serca [121]. Te badania wskazują, że modulacja nadaktywności układu endokannabinoidowego jest obiecującym podejściem terapeutycznym w leczeniu deficytów poznawczych związanych z zespołami genetycznymi.
Molekuły Modulujące Układ Endokannabinoidowy
Konopie, będące roślinnym lekarstwem, są złożoną mieszaniną kilku związków, w tym fenoli kannabinoidowe, fenole niekannabinoidowe (proste fenole, spiro-indany, dihydrofenantreny i dihydrostilbeny), flawonoidy, terpenoidy, alkohole, aldehydy, n-alkany, estry woskowe, steroidy i alkaloidy. W 1899 roku Wood wyizolował kannabinol (CBN), pierwszy związek oczyszczony z tej rośliny. Obecnie wyizolowano i zgłoszono ponad 500 różnych substancji pochodzących z roślin konopi należących do różnych klas, spośród których klasa związków kannabinoidowych jest najbardziej reprezentatywna, ponieważ ma ponad 120 zidentyfikowanych związków, takich jak delta-ośmiotetrahydrokannabinol (Δ8-THC) i delta-dziewięciotetrahydrokannabinol (Δ9-THC), CBD i CBN (Figura 6). Zidentyfikowano różne klasy metabolitów wtórnych z różnych części rośliny, z szerokim zakresem zastosowań (nutraceutyki, kosmetyki, aromaterapia i farmakoterapia), korzystnych dla ludzi. Jednak w przeszłości badania skupiały się na dwóch najbardziej obfitych fitokannabinoidach, THC i CBD, co skutkowało większą wiedzą na temat ich działań farmakologicznych i wzrastającym zainteresowaniem licznych możliwości działań leczniczych tej rośliny [109,122,123,124,125,126,127,128]. CBD zyskuje na znaczeniu w badaniach farmakologicznych od lat 70. XX wieku. Epidiolex®, oczyszczony doustny lek zawierający CBD, jest obecnie zatwierdzony przez Agencję Żywności i Leków (FDA) w Stanach Zjednoczonych do leczenia trudnych do leczenia napadów padaczkowych rozpoczynających się w dzieciństwie [129,130].
Rysunek 6. Struktury chemiczne głównych farmakologicznie aktywnych związków kannabinoidowych wyizolowanych z Cannabis sativa.
Jednakże biochemiczne podstawy działania farmakologicznego kannabinoidów pozostawały tajemnicą przez wiele lat. Wysoce lipofiliczna struktura molekularna kannabinoidów sugeruje, że wywierają one swoje działanie poprzez przenikanie błon komórkowych i oddziaływanie w ośrodkowym układzie nerwowym. Obecnie dostępne są istotne informacje na temat właściwości fizykochemicznych kannabinoidów. Nowoczesne selektywne ligandy dla receptorów kannabinoidowych mogą zawierać określone grupy substytuentów, które zwiększają kinetykę wiązania i zmniejszają skutki uboczne.
5. System endokannabinoidowy staje się obiecującym celem farmakoterapii przewlekłego bólu i zaburzeń nastroju.
Ból jest opisywany jako nieprzyjemne doświadczenie sensoryczne i emocjonalne związane z rzeczywistym lub potencjalnym uszkodzeniem tkanek lub w terminach takiego uszkodzenia. Kiedy ból utrzymuje się lub nawraca przez dłużej niż trzy miesiące, definiuje się go jako ból przewlekły, co ma znaczny wpływ na społeczeństwo. Szacuje się, że około 20% światowej populacji cierpi na ból przewlekły. Ważnym elementem jest również obserwacja depresji i lęku u takich pacjentów. Terapia bólu obejmuje zarówno leczenie farmakologiczne, jak i niefarmakologiczne. Do leczenia przewlekłego bólu często zaleca się leki przeciwdepresyjne, przeciwpadaczkowe oraz leki oddziałujące na układ nerwowy centralny. Substancje terapeutyczne uważane są za leki adiuwantowe, które pierwotnie nie zostały opracowane jako leki przeciwbólowe, ale wykazują właściwości przeciwbólowe i są lekami pierwszego rzutu w leczeniu bólu neuropatycznego i problemów psychiatrycznych. Niemniej jednak niektórzy pacjenci nie odczuwają ulgi w bólu i szukają innych terapii. Użycie ekstraktów z rośliny konopi (Cannabis sativa) do wywoływania różnych efektów leczniczych opisano już w trzecim tysiącleciu p.n.e. w chińskich tekstach. Ostatnio zainteresowanie właściwościami leczniczymi Cannabis sativa odrodziło się wraz z pojawieniem się systemu endokannabinoidowego (eCB), oferującego nie tylko nowe spojrzenie na mechanizmy działania terapeutycznego molekuł podobnych do kannabinoidów i fitokannabinoidów, ale także nowe molekularne cele dla farmakoterapii bólu.
CB1R może być aktywowany przez THC, co powoduje analgezję i wystąpienie działań niepożądanych (np. ból głowy, drętwienie, kaszel, uczucie pieczenia, zawroty głowy, uczucie odurzenia, senność, suchość w ustach i oczach). Wiele z tych zjawisk psychoaktywnych zależy od stężeń THC i może stanowić istotne wady jego stosowania w terapii farmakologicznej. Poprzednie badanie wykazało, że aktywacja CB1R powoduje upośledzenie pamięci. Ponadto THC i inne kannabinoidy aktywują receptory serotoniny 2A (5-HT2AR), wpływając na upośledzenie pamięci, działanie przeciwlękowe i interakcję społeczną. Badania wykazały, że CB1R i 5-HT2AR tworzą heteromery, które są wyrażane i funkcjonalnie aktywne w konkretnych obszarach mózgu zaangażowanych w upośledzenie pamięci, takich jak hipokamp i kora przedczołowa. Jednakże upośledzenie pamięci i lęk zostały zniesione u myszy typu dzikiego za pomocą antagonisty 5-HT2AR lub selektywnego zakłócenia heteromerów CB1R/5-HT2AR poprzez infuzję syntetycznych interferencyjnych peptydów, nie tracąc jednocześnie efektu przeciwbólowego. Trwające badania nad stosowaniem Cannabis pokazują, że sprzyja ono złagodzeniu bólu i rozłącza upośledzenie pamięci, co zmniejsza wady stosowania kannabinoidów jako substancji terapeutycznych. CB2R odgrywa istotną rolę w modulacji analgezji poprzez dwie ścieżki. Pierwszy mechanizm zachodzi w obwodowym układzie immunologicznym, gdzie receptory CB2 mediują analgezję, modyfikując profil cytokin i zapobiegając niepożądanym efektom w ośrodkowym układzie nerwowym. Po drugie receptory CB2 obecne w komórkach glejowych i neuronach przyczyniają się do złagodzenia bólu. Dodatkowo badania wykazały, że selektywne agonisty CB2 promują antynocicepcję. Ponadto badania nad ludźmi wykazały, że naturalne i syntetyczne kannabinoidy, takie jak dronabinol i nabilon, mają zastosowanie w łagodzeniu przewlekłego bólu, a zaobserwowano terapeutyczną skuteczność w leczeniu bólu i poprawie jakości życia pacjentów. System eCB jest rozpowszechniony w obrębie rdzenia kręgowego i nadrdzenia, dzięki czemu może skutecznie regulować przetwarzanie nocicepcyjne.
Lęk i zaburzenia paniki, główne stany depresyjne oraz choroba afektywna dwubiegunowa (choroba maniakalno-depresyjna) to poważne i potencjalnie zagrażające życiu komplikacje nastroju. Ponad 20% dorosłej populacji doświadcza zaburzeń nastroju w pewnym okresie życia. W ciągu ostatnich dziesięcioleci dokonano wielu postępów w leczeniu zaburzeń nastroju. Około 30% populacji nie reaguje na obecne terapie, dlatego trwa poszukiwanie nowych podejść farmakologicznych. Efekty psychoaktywne Cannabis obejmują działanie uspokajające, przeciwlękowe, nasenne i euforyczne. Niektóre z tych czynników pozytywnie wpływają na nastrój. Jednak u niektórych osób mogą pojawić się objawy takie jak paranoja, drażliwość, dysforia, depresja, depersonalizacja i brak motywacji, jako skutki uboczne. Reakcje te zależą od aktywności endokannabinoidowej pacjenta, dawki używanej (normalnie działającej stymulująco przy niskiej dawce i hamująco przy wysokiej dawce), proporcji fitokannabinoidów i składu terpenoidowego. Narasta dowody na rolę systemu endokannabinoidowego w regulacji nastroju. Badania kliniczne wykazały zaburzenia sygnalizacji endokannabinoidowej u pacjentów psychiatrycznych. Polimorfizmy genetyczne w CB1R i CB2R są związane z depresją, chorobą afektywną dwubiegunową oraz opornością na terapię, co zaobserwowano u pacjentów z depresją posiadających polimorfizm jednonukleotydowy w CB1R. Ponadto system endokannabinoidowy może modyfikować funkcje wszystkich osi podwzgórzowo-przysadkowo-nadnerczowej za pośrednictwem CB1R, a przewlekły stres wydaje się obniżać zdolność systemu endokannabinoidowego do hamowania stresu, co może prowadzić do psychopatologii, w tym depresji i lęku. W tym kontekście ważne jest przypomnienie losów leku rimonabantu, pierwszego blokera receptorów kannabinoidowych zatwierdzonego do leczenia zespołu metabolicznego, otyłości i palenia tytoniu. Jednak ze względu na ważne działania niepożądane, które ujawniły się u pacjentów po długotrwałym stosowaniu rimonabantu, firma Sanofi-Aventis wycofała go z rynku. Lek ten głównie wywiera korzystne efekty, blokując receptory CB1 na obwodzie. Jednak ze względu na swoją lipofilową naturę rimonabant może przenikać przez barierę krew-mózg i dostawać się do ośrodkowego układu nerwowego, co związane jest z rozwojem depresji, myśli samobójczych i zaburzeń lękowych. Właściwości podnoszące nastrój kannabinoidów są od dawna znane i uważane za nieszkodliwe. Niektóre składniki Cannabis lub ich mieszaniny mogą wykazywać działanie przeciwdepresyjne i/lub przeciwlękowe. Wielu pacjentów niereagujących na standardowe leki przeciwdepresyjne może skorzystać z leczenia medycznego Cannabis. Kannabinoidy mogą mieć potencjał terapeutyczny zarówno w depresji, jak i chorobie afektywnej dwubiegunowej. Dotyczy to pacjentów z zaburzeniami nastroju, którzy dodają Cannabis do istniejącego leczenia, ponieważ taka kombinacja może poprawić skuteczność leku i/lub zmniejszyć jego skutki uboczne. Cannabis może działać stabilizująco na nastrój w chorobie afektywnej dwubiegunowej i pełnić funkcję adiuwantową w leczeniu litu. Pacjenci z zaburzeniem nastroju mogą nie być obiektywni w ocenie swojego stanu i nie mogą decydować samodzielnie o modyfikacji leczenia. Dlatego ważna jest profesjonalna opieka i kontrola.
6. Harmonizacja układu endokannabinoidowego poprzez praktyki zdrowia integracyjnego i uzupełniającego (ICHP).
Integracyjne i uzupełniające praktyki zdrowia (ICHP), znane również jako „komplementarne i alternatywne metody leczenia (CAM)”, obejmują różne systemy medyczne i zdrowotne, praktyki i produkty, które obecnie nie są częścią konwencjonalnych terapii. CAM jest podzielony na trzy główne grupy: „produkty naturalne” (suplementy diety i ziołowe), „medycyna umysłowo-cielesna” (medytacja, joga i akupunktura) oraz „praktyki oparte na ciele” (masaż i manipulacja kręgosłupa) [195,196].
Badanie na gryzoniach przeprowadzone przez Chena i in. pokazało, że elektroakupunktura promuje aktywność przeciwnocyceptywną u zwierząt i zwiększa poziomy AEA w tkance skórnej. Stwierdzono również, że efekty przeciwnocyceptywne zostały osłabione przy użyciu AM630, antagonisty CB2R, ale nie przy użyciu antagonisty CB1R AM25 [197]. Ponadto AEA zwiększyło ekspresję CB2R w skórze [198,199]. Aktywacja CB2R w skórze prawdopodobnie stymuluje uwalnianie β-endorfiny, która następnie działa na obwodowe receptory μ-opioidowe, hamując przeciwnocycepcję [51]. Ponadto elektroakupunktura udzielała ochrony neurologicznej przed niedokrwieniem mózgu poprzez stymulację mobilizacji endokannabinoidów w mózgu i aktywację CB1R [200,201].
Sadhasivam i in. sugerują, że endokannabinoidy mogą pełnić rolę biomarkerów po sesji medytacyjnej. Wyniki sugerują, że główne endokannabinoidy, w tym AEA, 2-AG, 1-AG, DEA, OLA oraz czynnik neurotropowy pochodzenia mózgowego (BDNF), zwiększały się po medytacji u ponad 70% pacjentów, co wskazuje na istotną rolę tych biomarkerów w mechanizmie działania medytacji [202]. Badania sugerują istnienie korelacji między akupunkturą a układem endokannabinoidowym poprzez dzielenie się efektami biologicznymi, takimi jak analgezja, neuroprotekcja i regulacja układu sercowo-naczyniowego. Lepsze zrozumienie tych wewnętrznych powiązań między akupunkturą a CES może pozwolić na opracowanie nowych technik łączących akupunkturę z substancjami terapeutycznymi ukierunkowanymi na sygnał lizy endokannabinoidów [203,204,205,206].
Inne badanie wykazało, że masaż i osteopatyczna manipulacja u zdrowych uczestników zwiększyły poziomy AEA we krwi o 168% w porównaniu z poziomami przed leczeniem; nie stwierdzono zmian w poziomach 2-AG. U uczestników poddanych manipulacji pozorowanej (kontrola) nie zaobserwowano zmian [207]. Zintegrowane podejście, łączące akupunkturę, masaż, jogę, metody umysłowo-cielesne i medyczną konopię, może być skuteczne. Na przykład pacjent z przewlekłym bólem neuropatycznym wykazuje poprawę w obrazie klinicznym, gdy jest leczony w ten sposób [208]. W związku z tym konieczne jest zastosowanie złożonego, zindywidualizowanego podejścia, podkreślając prowadzenie pacjenta i zaangażowanie w metody integracyjne oraz lecznicze stosowanie konopi.
7. Perspektywy badawcze i trendy w układzie endokannabinoidowym.
Od początku badań naukowych nad kannabinoidami, ze szczególnym uwzględnieniem izolacji i identyfikacji fitokannabinoidów, takich jak THC, naukowcy nieustannie doskonalą wiedzę na temat farmakologii, biochemii i klinicznych efektów Cannabis. Od lat dobrze znane są efekty fizjologiczne jej spożycia, zwłaszcza stan euforii. Jednak to, co zachodzi wewnątrz naszych organizmów na poziomie molekularnym, zwłaszcza w mózgu, aby zmienić świadomość, wciąż pozostaje nieznane. W 1973 roku amerykańscy badacze zidentyfikowali receptory w mózgu związane z opioidami. Niektórzy naukowcy spodziewali się, że odkrycie receptorów dla marihuany nastąpi szybko. Jednakże nie było to takie łatwe ani szybkie, jakby chcieli. Badania przeprowadzone przez Allyn Howletta i Williama Devane’a wykazały, że receptory kannabinoidowe są obecne w mózgu bardziej niż jakiekolwiek inne receptory sprzężone z białkami G [206,209,210,211].
CB1R i CB2R, będące częścią układu endokannabinoidowego, odgrywają kluczową rolę w licznych warunkach fizjologicznych i chorobach ludzkich. Dlatego podejmowane są znaczne wysiłki w celu opracowania ligandów dla CB1R i CB2R, co skutkuje powstaniem setek fitokannabinoidów i syntetyków, które wykazały zróżnicowane powinowactwo do leczenia różnych chorób [17]. Jednak tylko kilka z tych ligandów jest używanych klinicznie. Obecnie dzięki krioelektronowej mikroskopii ujawniono bardziej szczegółowe informacje strukturalne receptorów kannabinoidowych, co przyspieszyło odkrywanie substancji opartych na strukturze [209]. Jednocześnie pojawiły się nowe kannabinoidy peptydopodobne pochodzenia zwierzęcego, które wykazują potencjalne efekty terapeutyczne in vivo na receptorach kannabinoidowych [212,213].
Z perspektywy produktów naturalnych oczekuje się, że nowe kannabinoidy zostaną odkryte i uznane za prototypy obiecujących leków pochodzących z różnych źródeł i gatunków naturalnych, takich jak jady zwierzęce, które stanowią prawdziwą farmakopeę toksyn modulujących różne cele, w tym kanały jonowe i receptory sprzężone z białkami G, takie jak CB1R i CB2R, z istotnym powinowactwem i selektywnością [214]. Dlatego sądzi się, że odkrywanie nowych kannabinoidów poprzez badanie bioróżnorodności gatunków zamieszkujących Ziemię to obszar, który jeszcze nie został zbadany.
8. Wnioski
W tej recenzji omówiono role receptorów kannabinoidowych i ich agonistów w wielu stanach chorobowych. Od czasu rozpoczęcia badań nad pochodnymi Cannabis i biologicznymi funkcjami izolowanych związków w doświadczalnych i ludzkich chorobach wykazują one obiecujące wyniki. Wydaje się, że selektywne ligandy określonych receptorów kannabinoidowych mogą wywoływać korzystne rezultaty w zależności od warunków klinicznych. Należy zachęcać do prowadzenia dalszych badań nad biologiczną funkcją każdej pochodnej Cannabis.
Receptory kannabinoidowe typu 1 (CB1) zostały wcześniej wykryte w komórkach β trzustki, gdzie wpływają na działanie insuliny. Teraz raportujemy, że receptory CB1 tworzą kompleks heteromeryczny z receptorami insuliny i Gαi, co hamuje aktywność kinazy receptora insuliny w komórkach β poprzez bezpośrednie wiązanie z pętlą aktywacyjną w domenie kinazy tyrozynowej receptora insuliny. W rezultacie fosforylacja proapoptotycznego białka Bad została zmniejszona, prowadząc do aktywacji Bad i indukcji śmierci komórek β. Farmakologiczne blokowanie lub niedobór genetyczny receptorów CB1 prowadziły do obniżenia poziomu glukozy we krwi i zwiększenia przeżycia komórek β po uszkodzeniu z powodu zintensyfikowanej sygnalizacji receptora insuliny i zmniejszonej aktywacji Bad. Te wyniki dostarczają bezpośrednich dowodów na fizyczne i funkcjonalne interakcje między receptorami CB1 a insulinowymi oraz przedstawiają mechanizm, dzięki któremu antagonisty receptorów CB1 działające obwodowo poprawiają działanie insuliny w tkankach wrażliwych na insulinę, niezależnie od innych efektów metabolicznych receptorów CB1.
Cel przeglądu: Otyłość brzuszna jest ściśle związana z cukrzycą typu 2 i nadaktywnością układu endokannabinoidowego. Niniejszy przegląd ma na celu ocenę roli układu endokannabinoidowego w dysregulacji glukozy oraz wpływu blokady receptora kannabinoidowego typu 1 na metabolizm glukozy zarówno w modelach zwierzęcych, jak i u ludzi z nadwagą/otyłością, zwłaszcza z cukrzycą typu 2.
Ostatnie odkrycia: Receptory kannabinoidowe typu 1 zostały zidentyfikowane nie tylko w mózgu, ale także w tkance tłuszczowej, jelitach, wątrobie, mięśniach szkieletowych, a nawet trzustce, wszystkich narządach odgrywających kluczową rolę w metabolizmie glukozy i cukrzycy typu 2. Rimonabant, pierwszy selektywny bloker receptora kannabinoidowego typu 1 stosowany klinicznie, wykazał skuteczność w redukcji masy ciała, obwodu talii, glikowanej hemoglobiny, triglicerydów, wskaźnika oporności na insulinę oraz zwiększeniu stężeń cholesterolu HDL i adiponektyny u pacjentów z cukrzycą typu 2, potwierdzając dane dotyczące pacjentów z nadwagą/otyłością, którzy nie mieli cukrzycy. Prawie połowa zmian metabolicznych, w tym redukcja glikowanej hemoglobiny, nie mogła być wyjaśniona tylko utratą wagi, zgodnie z bezpośrednimi efektami obwodowymi.
Podsumowanie: Blokada receptora kannabinoidowego typu 1 redukuje spożycie pokarmu i masę ciała, poprawiając regulację metaboliczną poza samą utratą wagi. Ze względu na pozytywny wpływ na metabolizm glukozy, rimonabant zasługuje na uwzględnienie w leczeniu pacjentów z nadwagą/otyłością i cukrzycą typu 2.
źródło: https://pubmed.ncbi.nlm.nih.gov/18542014/
Zarządzaj zgodami plików cookie
Używamy plików cookies w celu optymalizacji naszej witryny i naszych serwisów. Więcej informacji:
Funkcjonalne
Zawsze aktywne
Przechowywanie lub dostęp do danych technicznych jest ściśle konieczny do uzasadnionego celu umożliwienia korzystania z konkretnej usługi wyraźnie żądanej przez subskrybenta lub użytkownika, lub wyłącznie w celu przeprowadzenia transmisji komunikatu przez sieć łączności elektronicznej.
Preferencje
Przechowywanie lub dostęp techniczny jest niezbędny do uzasadnionego celu przechowywania preferencji, o które nie prosi subskrybent lub użytkownik.
Statystyka
Przechowywanie techniczne lub dostęp, który jest używany wyłącznie do celów statystycznych.Przechowywanie techniczne lub dostęp, który jest używany wyłącznie do anonimowych celów statystycznych. Bez wezwania do sądu, dobrowolnego podporządkowania się dostawcy usług internetowych lub dodatkowych zapisów od strony trzeciej, informacje przechowywane lub pobierane wyłącznie w tym celu zwykle nie mogą być wykorzystywane do identyfikacji użytkownika.
Marketing
Przechowywanie lub dostęp techniczny jest wymagany do tworzenia profili użytkowników w celu wysyłania reklam lub śledzenia użytkownika na stronie internetowej lub na kilku stronach internetowych w podobnych celach marketingowych.

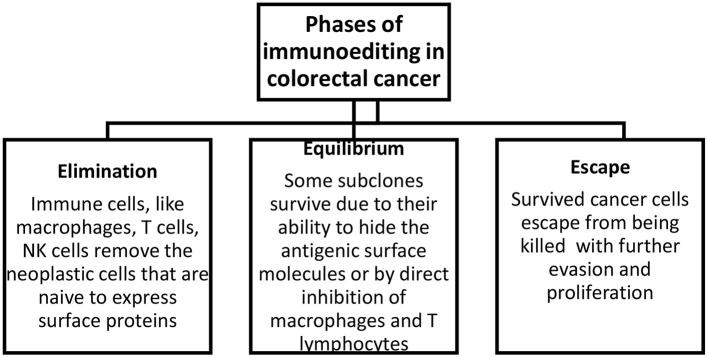

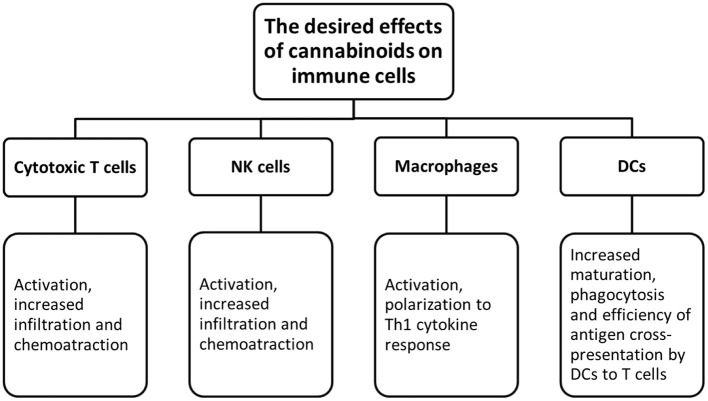
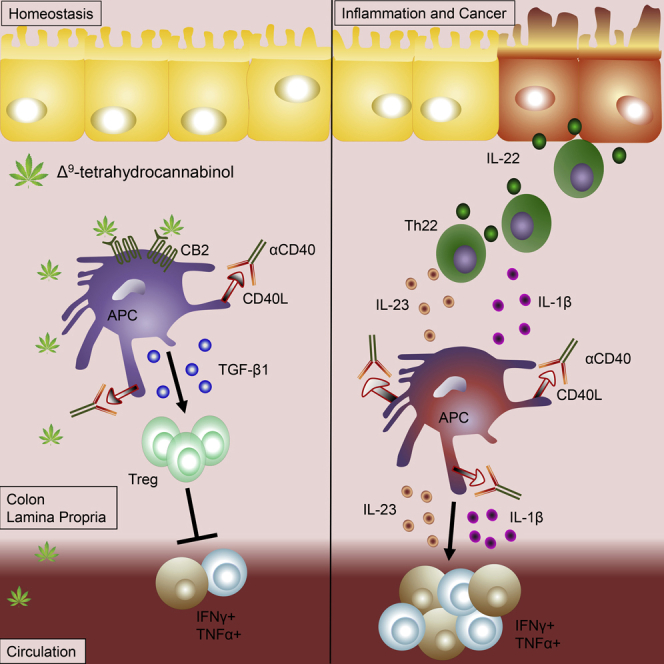
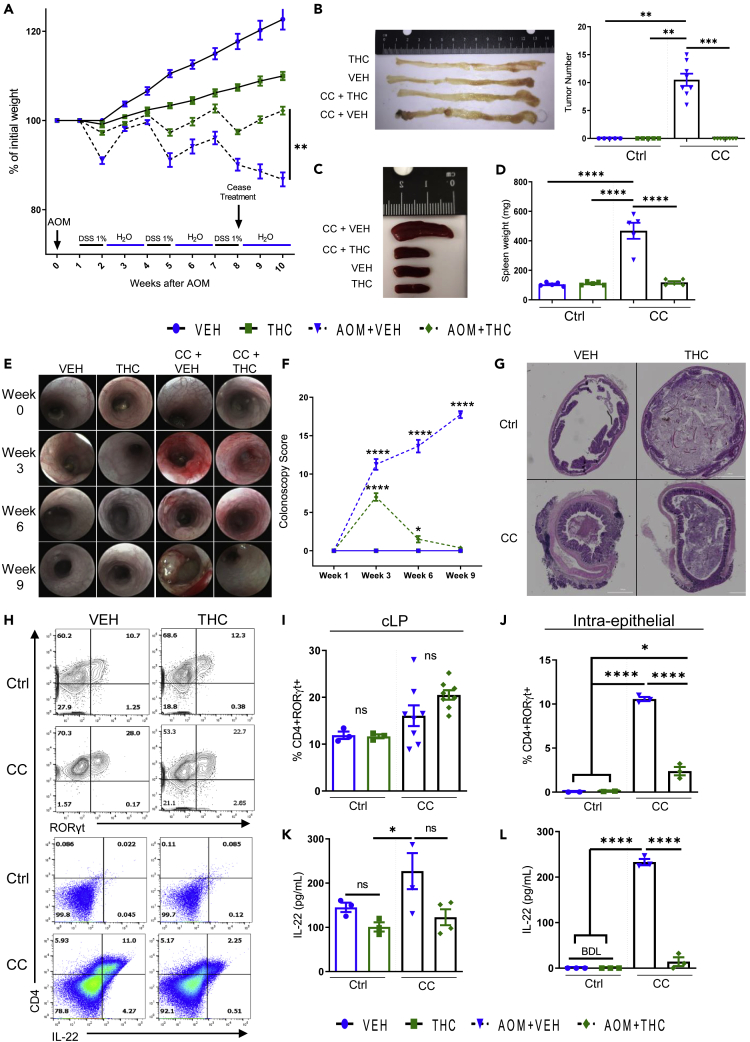
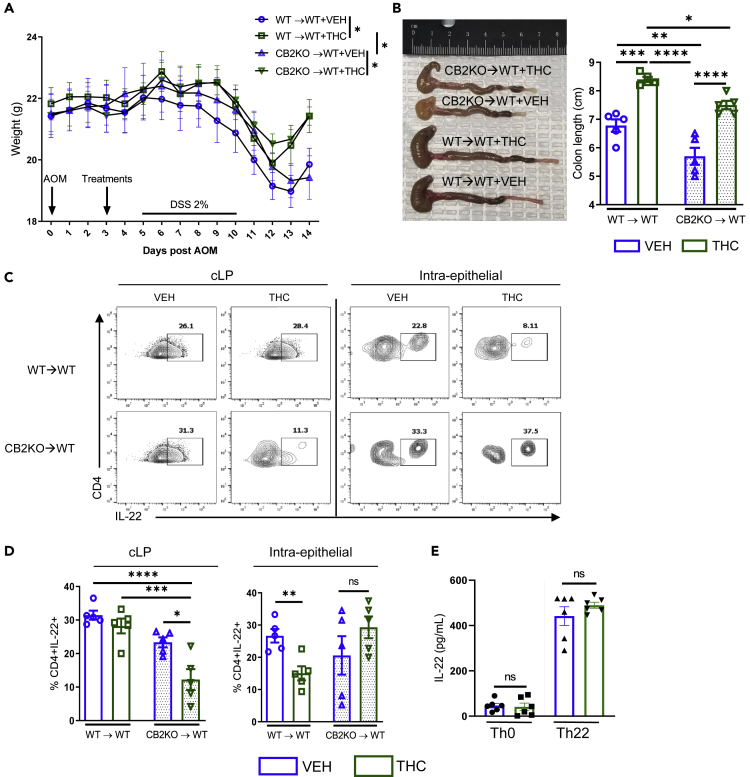
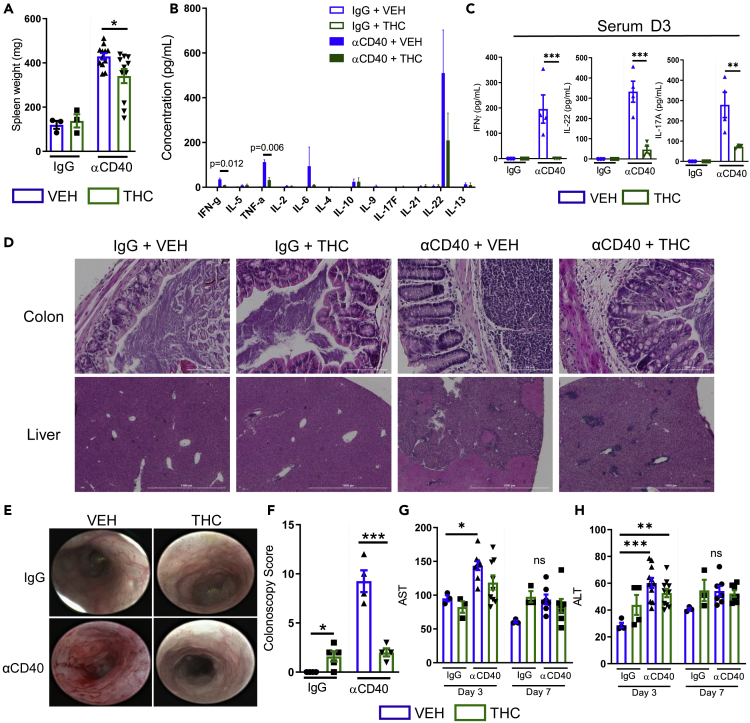
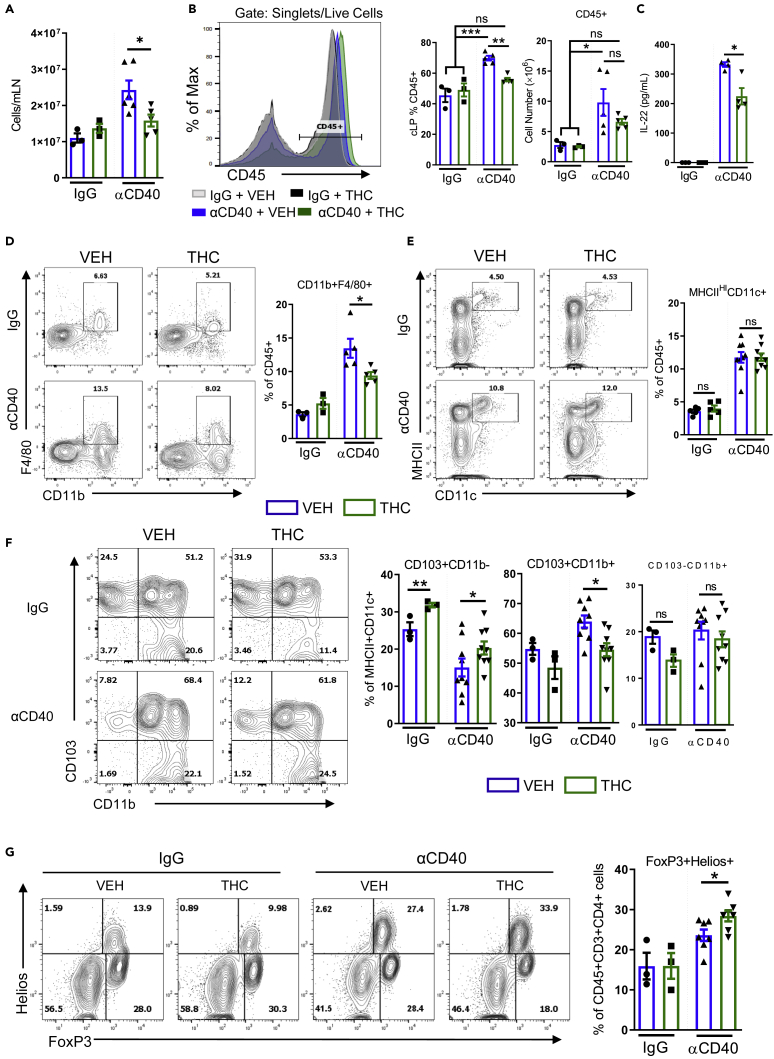
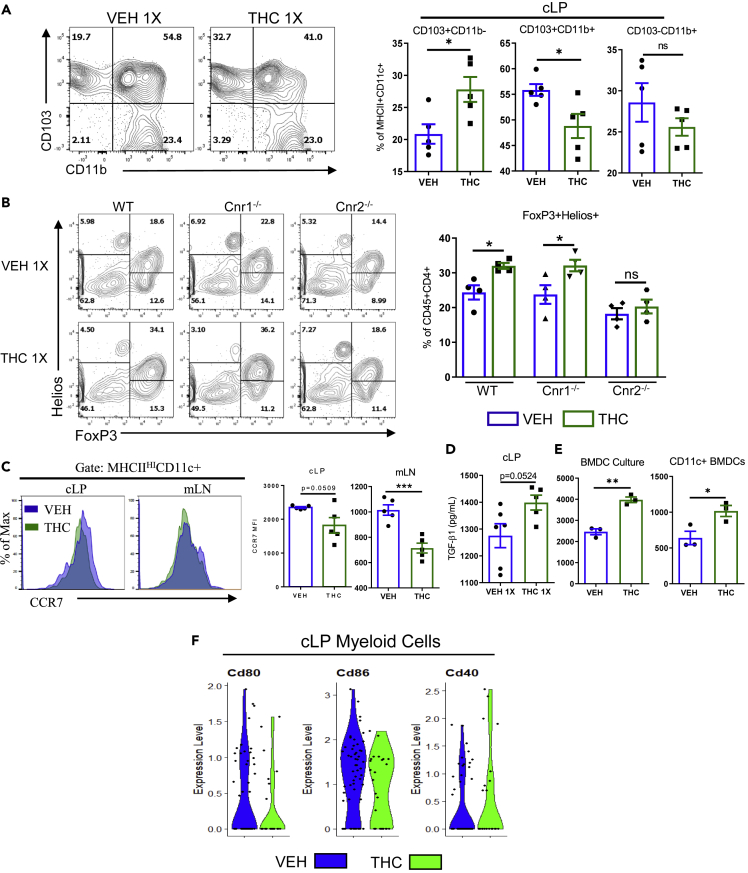
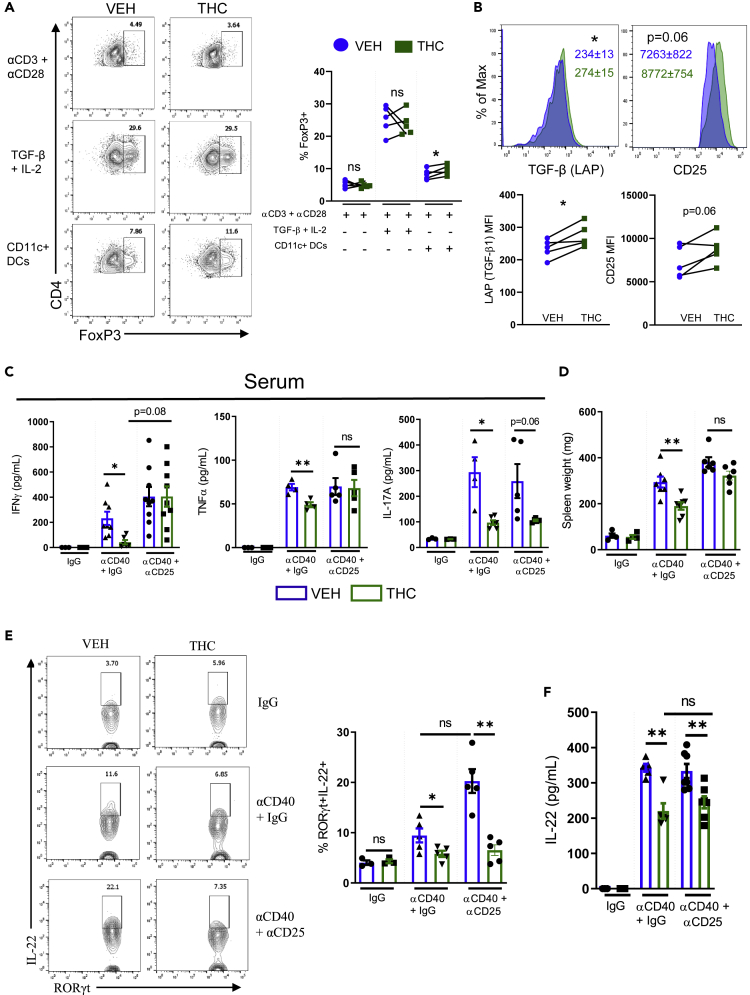
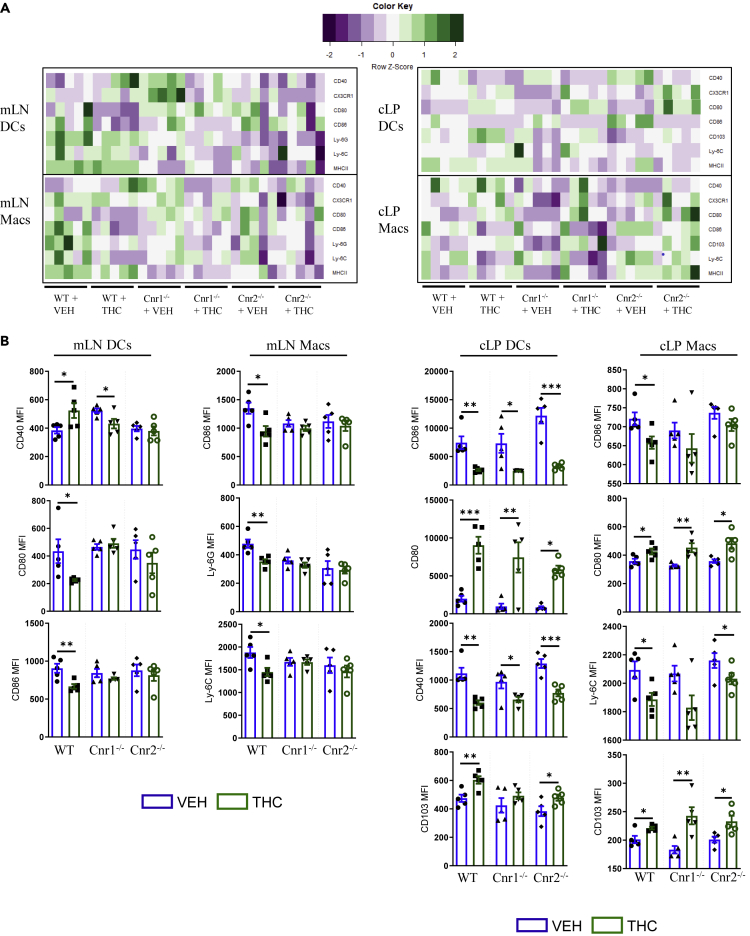
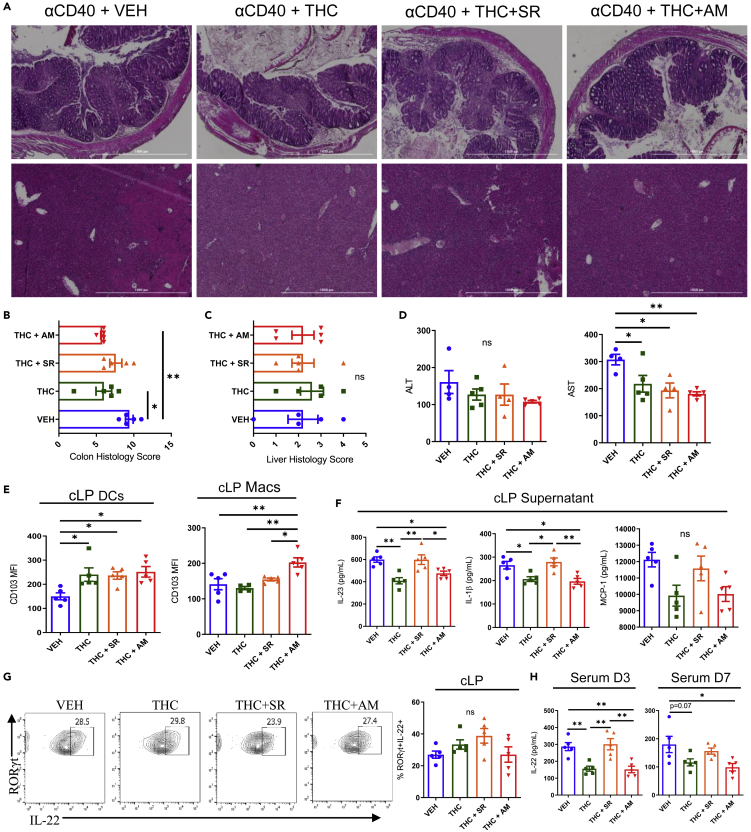
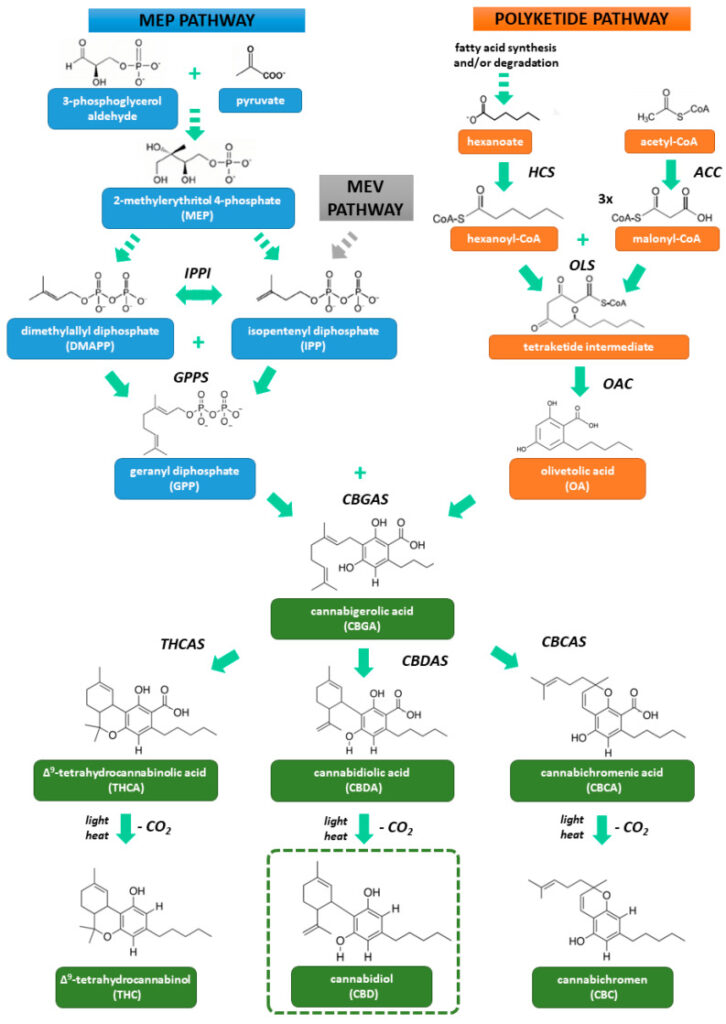
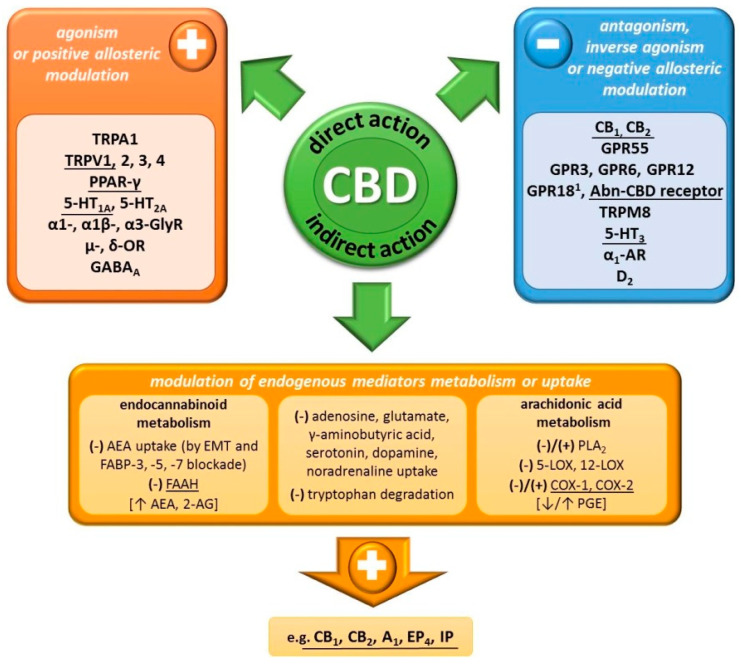
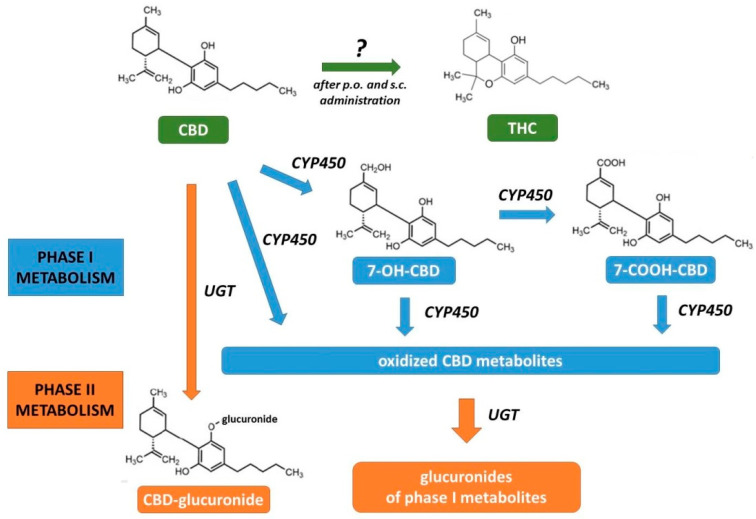
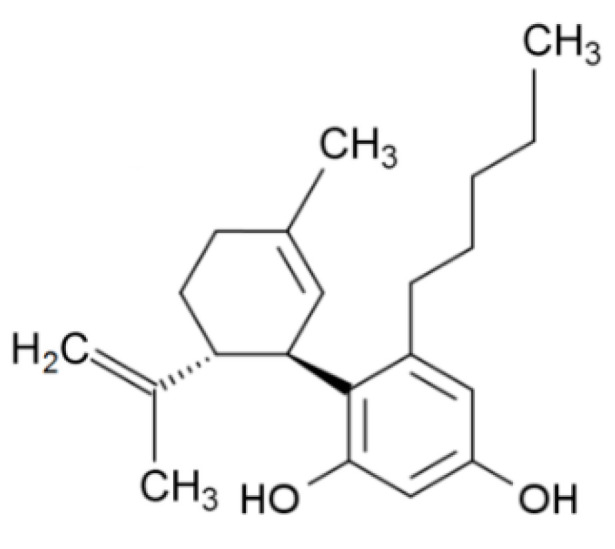
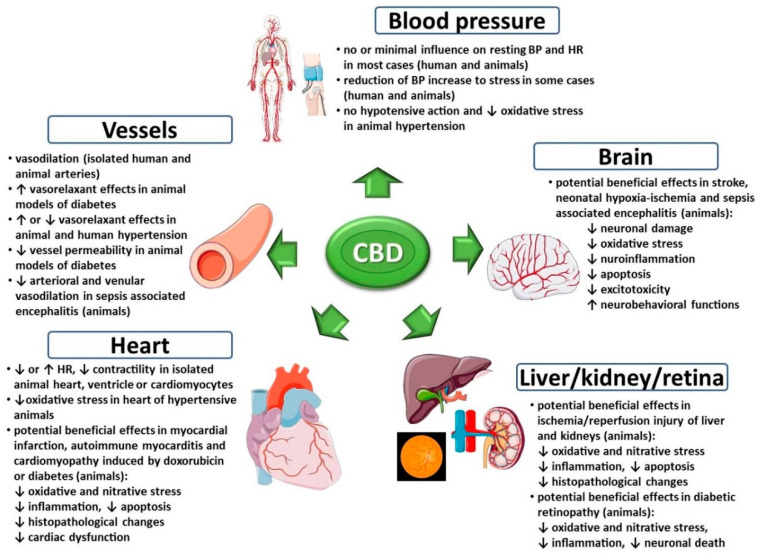
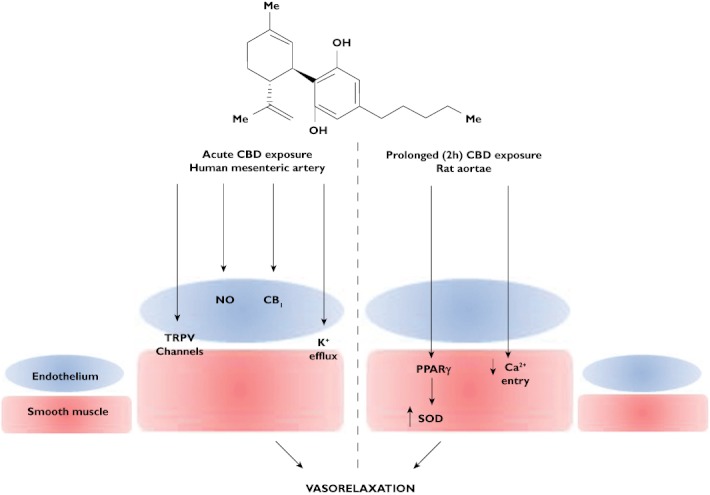
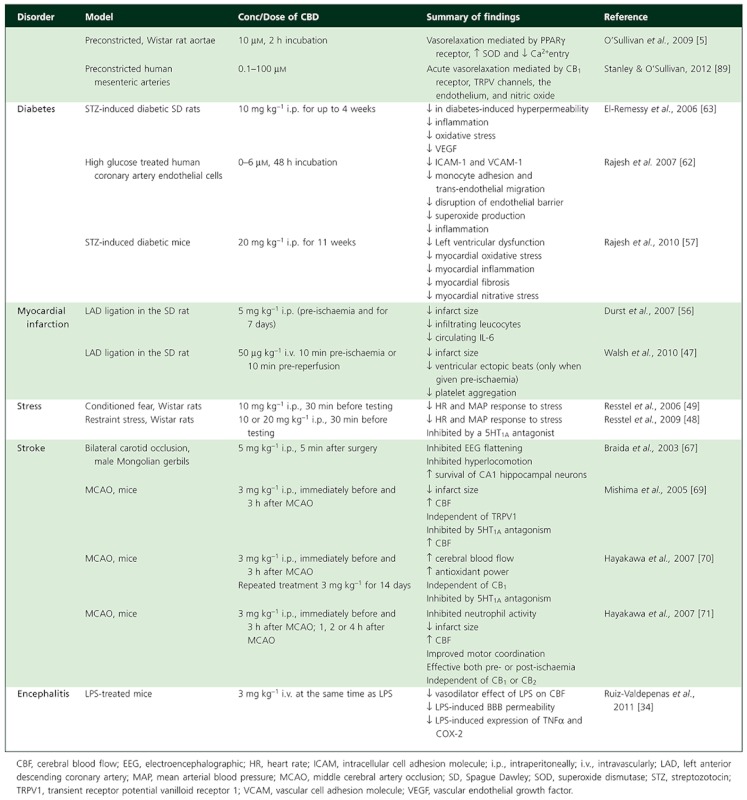
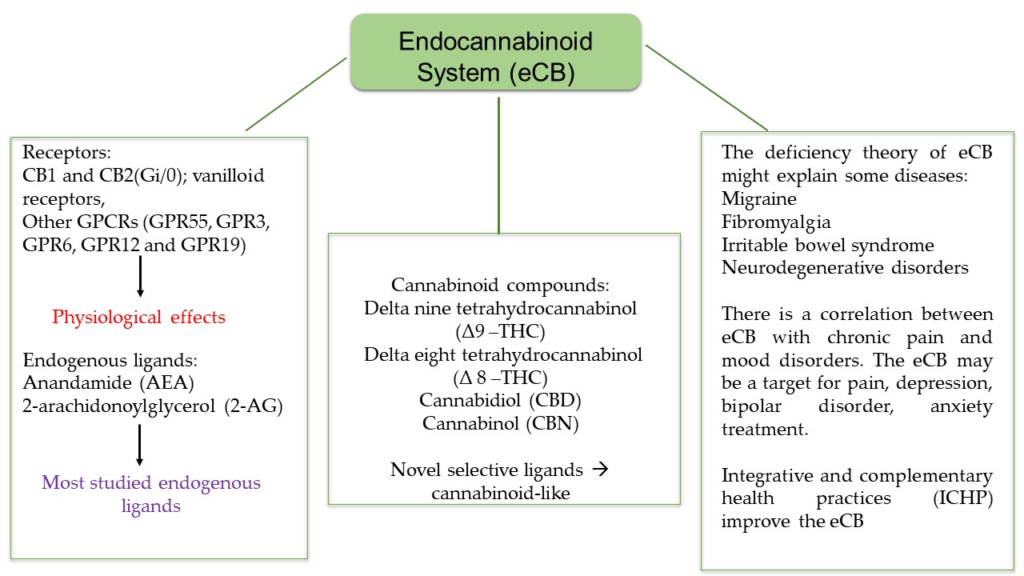
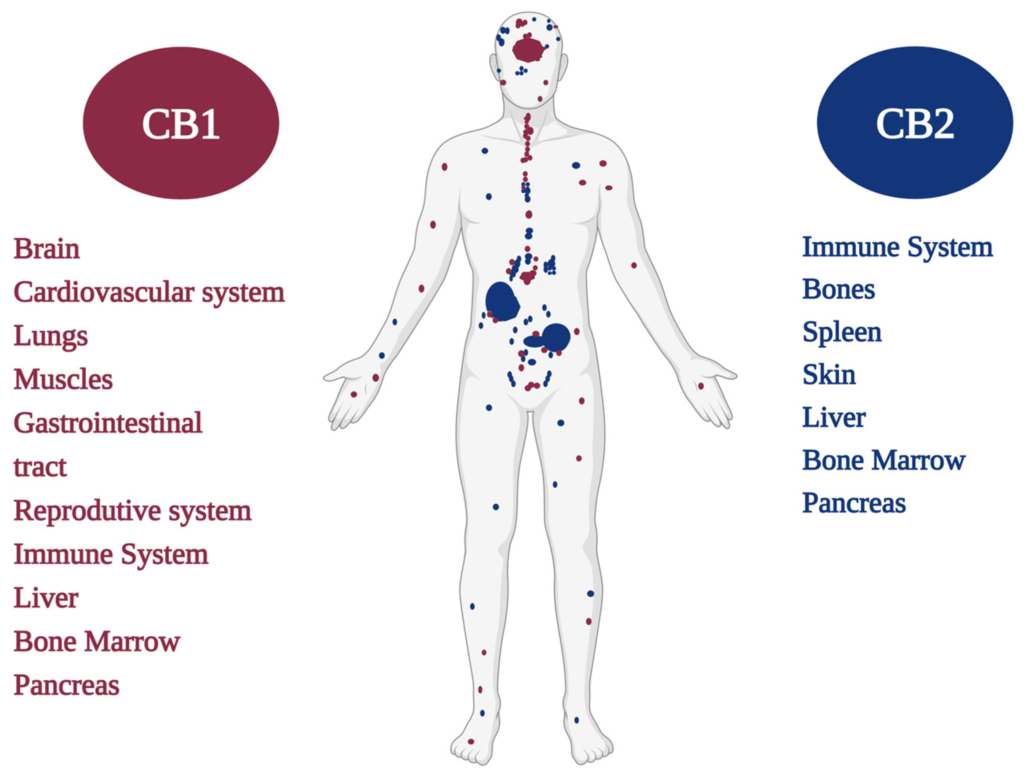
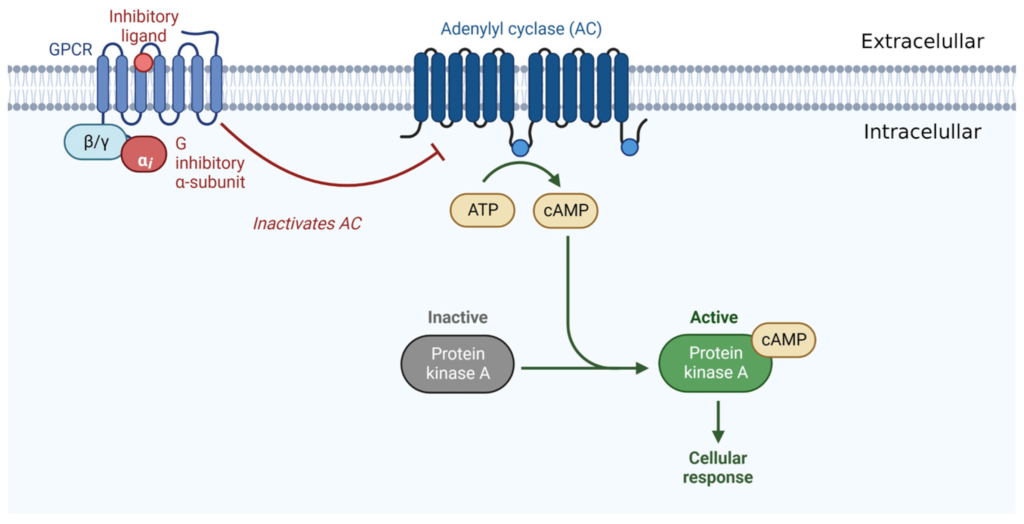
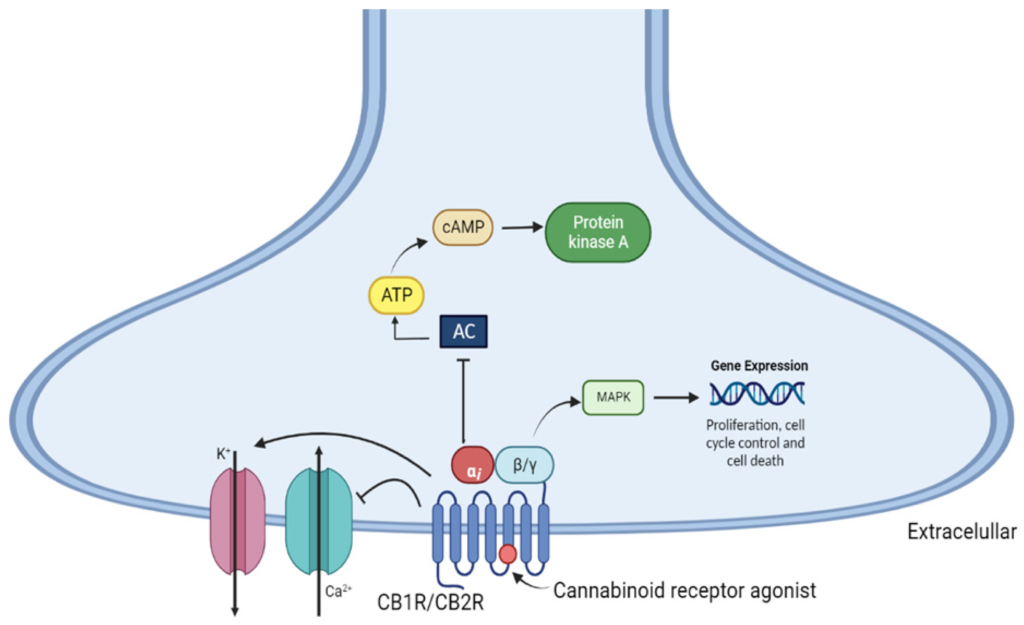
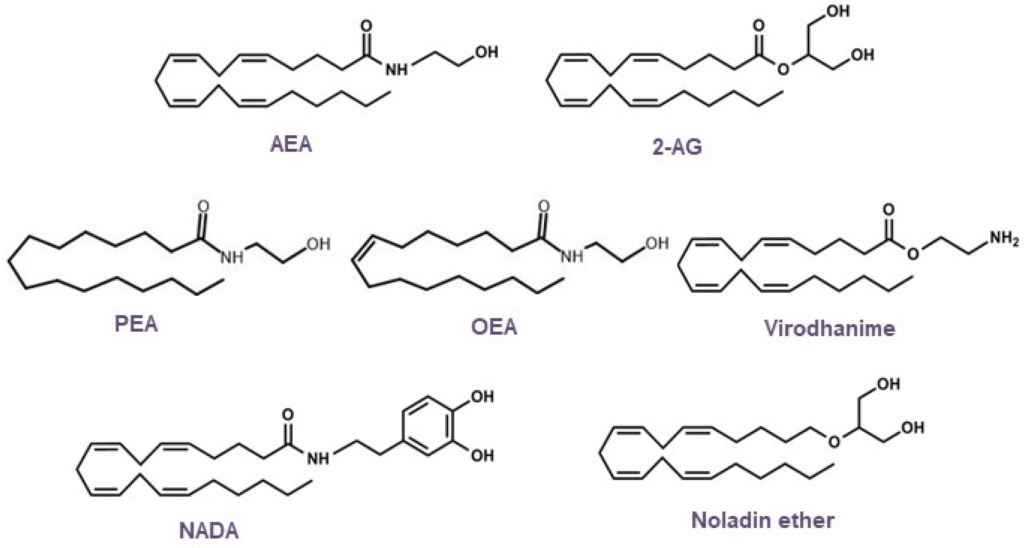
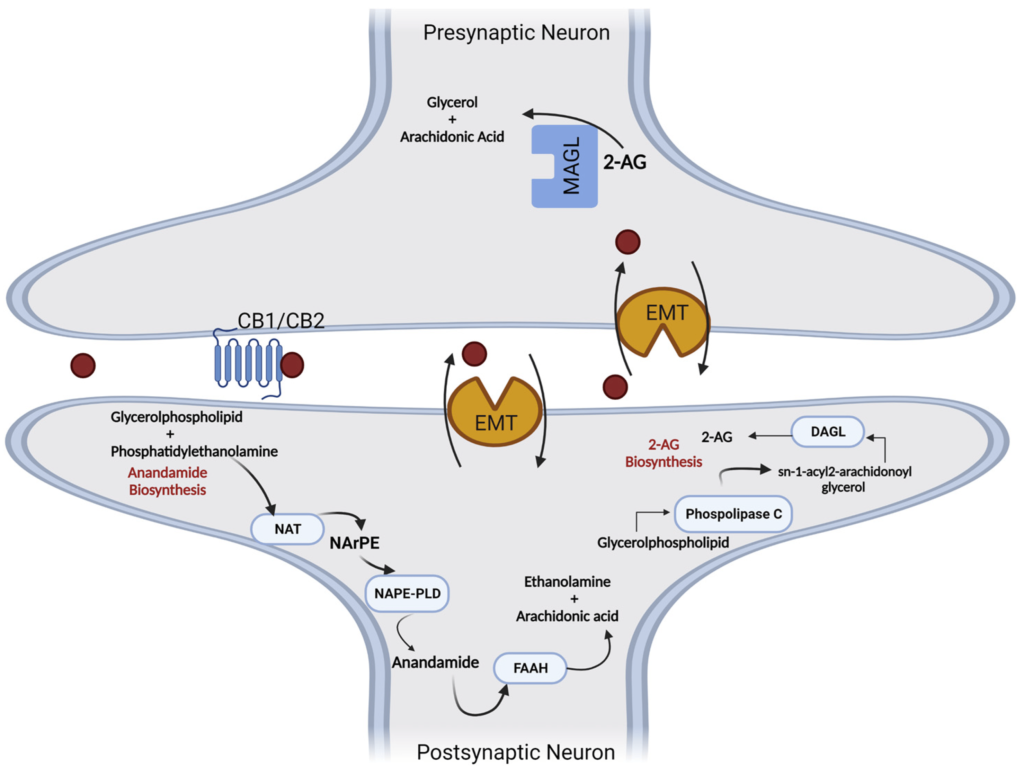
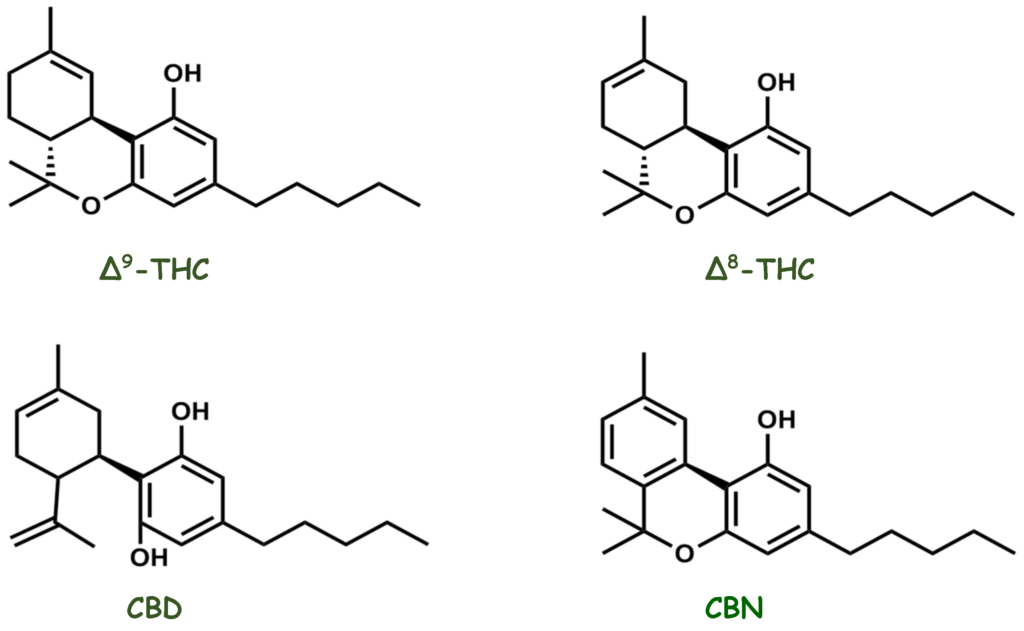
{kind=link}
{kind=link}