Cel przeglądu Przegląd ten identyfikuje i umieszcza w kontekście niedawno opublikowane artykuły, które posunęły do przodu zrozumienie funkcji komórek zrazikowych trzustki oraz mechanizmów regulujących te funkcje.
Ostatnie odkrycia Receptory obecne na komórkach zrazikowych, zwłaszcza receptory dla cholecystokininy i sekretyny, zostały lepiej scharakteryzowane pod względem molekularnego charakteru interakcji ligand-receptor. Inne doniesienia opisują potencjalną regulację komórek zrazikowych przez GLP-1 i kannabinoidy. Sygnalizacja wapniowa wewnątrzkomórkowa pozostaje w centrum sprzężenia bodziec-sekrecja, a jej regulacja została bardziej zdefiniowana. Niedawne badania zidentyfikowały konkretne kanały przekazujące uwolnienie wapnia z magazynów wewnątrzkomórkowych i napływ przez błonę komórkową. Prace na etapie poniżej mediatorów wewnątrzkomórkowych skupiły się na molekularnych mechanizmach egzocytozy, zwłaszcza zaangażowanych w to małych białkach G, białkach SNARE i cząstkach szlaku sekrecyjnego. Oprócz sekrecji, niedawne badania dalsze zdefiniowały regulację wzrostu trzustki zarówno w adaptacyjnej regulacji diety i hormonów, jak i w regeneracji po uszkodzeniach trzustki. Śledzenie linii komórkowej zostało wykorzystane do pokazania wkładu różnych typów komórek. Ważność konkretnych aminokwasów jako cząsteczek sygnalizacyjnych aktywujących szlak mTOR jest wyjaśniana.
Podsumowanie Zrozumienie mechanizmów regulujących funkcję komórek zrazikowych trzustki przyczynia się do wiedzy na temat normalnej funkcji trzustki i zmian w przebiegu chorób.
Optymalna homeostaza glukozy wymaga precyzyjnej adaptacji liczby komórek beta trzustki wydzielających insulinę w wyspach trzustki. Insulina pozytywnie reguluje proliferację komórek beta w sposób autokrynny poprzez szlak sygnalizacyjny receptora insuliny (IR). Wychodzi teraz na jaw, że aktywacja/antagonizm receptora kannabinoidowego typu 1 (CB1R) wpływa na działanie insuliny w tkankach wrażliwych na insulinę. Niemniej jednak nie zostało jednoznacznie ustalone, na których komórkach wyrażane są CB1R i jakie pełnią funkcje w wyspach trzustki. W ramach niniejszego badania przeprowadziliśmy badanie mające na celu sprawdzenie, czy endogenne kannabinoidy (ECs) wewnątrzwyspowe regulują proliferację komórek beta oraz czy wpływają na działanie insuliny.
METODY BADAŃ
Zmierzyliśmy produkcję ECs w izolowanych ludzkich i mysich wyspach oraz w linii komórkowej beta w odpowiedzi na glukozę i KCl. Oceniliśmy obecność w pełni funkcjonującego systemu EC w ludzkich i mysich wyspach, kilku liniach komórkowych beta oraz myszach z brakiem receptora CB1R (CB1R−/−). Zbadaliśmy, czy ECs wpływają na fizjologię komórek beta poprzez regulację działania insuliny i wykazaliśmy potencjał terapeutyczny manipulacji systemem EC u myszy diabetycznych (db/db).
WYNIKI
ECs są wytwarzane wewnątrz komórek beta, które również wyrażają CB1Rs, działające w pełni, gdy są aktywowane przez ligandy. Blokada genetyczna i farmakologiczna CB1R prowadzi do zwiększenia sygnalizacji IR przez szlak białka podścieliska insuliny 2-AKT w komórkach beta, co skutkuje zwiększeniem proliferacji i masy komórkowej beta. Antagonizm CB1R u myszy db/db prowadzi do obniżenia stężenia glukozy we krwi, zwiększenia proliferacji i masy komórkowej beta, oraz zwiększonej sygnalizacji IR w komórkach beta. Ponadto aktywacja CB1R utrudnia autodfosforylację IR indukowaną insuliną w komórkach beta w sposób zależny od podjednostki Gαi.
WNIOSKI
Te wyniki stanowią bezpośrednie dowody na funkcjonalne oddziaływanie między CB1R a sygnalizacją IR zaangażowaną w regulację proliferacji komórek beta i stanowić będą podstawę do opracowania nowych interwencji terapeutycznych w celu poprawy funkcji i proliferacji komórek beta w cukrzycy.
Endokannabinoidy i receptory kannabinoidowe CB1 są znane z ogólnej roli w homeostazie energetycznej. Jednak badania kliniczne z pierwszą generacją blokerów CB1, obecnie wycofane z powodu skutków ubocznych psychiatrycznych, początkowo miały na celu zmniejszenie spożycia jedzenia i masy ciała, a nie czynników ryzyka metabolicznego związanego z otyłością. W tym przeglądzie omawiamy, w jaki sposób, oprócz promowania spożycia energii, endokannabinoidy kontrolują metabolizm lipidów i glukozy w kilku narządach obwodowych, szczególnie w wątrobie i tkance tłuszczowej. Pojawiają się także bezpośrednie działania w mięśniach szkieletowych i trzustce. Ta wiedza może pomóc w projektowaniu przyszłych terapii zespołu metabolicznego.
Cel: Nowe pochodne kwasu tetrahydrokannabinolowego (THCA) o nazwie ALAM027 i ALAM108 zostały zaproponowane do leczenia nowotworu trzustki.
Metody: Wpływ nowych kannabinoidów ALAM027 i ALAM108 został zbadany in vitro na liniach komórkowych PANC-1 i AsPC-1 za pomocą testu CellTiter Glo. Do badania in vivo aktywności przeciwnowotworowej tych związków na komórkach PANC-1 wykorzystano model przeszczepu nowotworu trzustki.
Wyniki: Badanie in vitro nowych kannabinoidów wykazało większą aktywność ALAM108 niż ALAM027 zarówno w przypadku komórek PANC-1, jak i AsPC-1. Badanie in vivo nowych kannabinoidów na komórkach PANC-1 wykazało, że ich doustne podawanie było skuteczne w zmniejszaniu objętości i masy guza, nie powodując przy tym żadnych dolegliwości ani utraty wagi u myszy.
Wnioski: Kannabinoidy ALAM108 i ALAM027 hamowały wzrost guza 1,6–2 razy u myszy z ludzkimi komórkami PANC-1.
Słowa kluczowe: AsPC-1, kannabinoidy, in vitro, in vivo, PANC-1, THCA.
Rak trzustki to jedna z najniebezpieczniejszych form nowotworów ze względu na jego agresywny wzrost, wczesne przerzuty i słabą odpowiedź na wszelkie znane metody terapeutyczne. Stanowi obecnie czwartą najczęstszą przyczynę zgonów z powodu raka, a przewiduje się, że do 2030 roku stanie się drugą najczęstszą przyczyną zgonów z powodu nowotworów, zaraz po raku płuc, co wymaga pilnych działań w zakresie nowych strategii terapeutycznych, które mogą poprawić wyniki leczenia tej choroby.
Ostatnio rosnące zainteresowanie przeciwnowotworową aktywnością kannabinoidów doprowadziło do licznych badań obejmujących coraz więcej rodzajów nowotworów. Naturalne i syntetyczne kannabinoidy wykazały zdolność wpływania na proliferację, migrację i apoptozę komórek nowotworowych poprzez bezpośrednie i pośrednie aktywowanie receptorów kannabinoidowych CB1 i CB2. W przypadku raka trzustki wykazano, że ilość ekspresji receptorów CB1 i CB2 w komórkach nowotworowych jest znacznie wyższa niż w komórkach normalnych, co otwiera drogę do wykorzystania przeciwnowotworowych zdolności kannabinoidów do eliminacji komórek nowotworowych bez wpływu na normalne tkanki trzustki.
Dotychczas aktywność przeciwnowotworową kannabinoidów wobec guza trzustki była badana tylko w kilku badaniach przedklinicznych, które wykazały obiecującą aktywność przeciwnowotworową tetrahydrokannabinolu (THC) oraz niektórych kannabinoidów syntetycznych, takich jak WIN 55,212-2. Jednak patrząc w przyszłość i przechodząc do etapów klinicznych, THC jest mało prawdopodobne, aby było stosowane w leczeniu guzów ze względu na jego wyraźny efekt psychoaktywny. Jednocześnie badanie kannabinoidów o podobnej strukturze, ale pozbawionych właściwości psychoaktywnych, jest niewątpliwie interesujące. Jednym z obiecujących kierunków może być modyfikacja naturalnego kwasu tetrahydrokannabinolowego (THCA) poprzez syntezę jego pochodnych w grupie karboksylowej. Chociaż sam THCA wykazuje niewielką aktywność przeciwnowotworową wobec raka trzustki, modyfikacja fragmentu karboksylowego jego cząsteczki wykazała znaczny wzrost in vitro hamowania wzrostu komórek. W związku z tym dalsze modyfikacje grupy karboksylowej THCA mogą prowadzić do związków o bardziej specyficznym działaniu na komórki guza trzustki. Celem tej pracy jest zbadanie nowych pochodnych THCA, ALAM027 i ALAM108, na komórkach nowotworowych trzustki PANC-1 i AsPC-1.
Endokannabinoidy przez aktywację swoistych receptorów wpływają na regulację homeostazy energetycznej ustroju. Kontrolują m.in. pobór pokarmu, wydzielanie insuliny, metabolizm lipidów i glukozy, magazynowanie tłuszczów. Głównym substratem energetycznym mięśnia sercowego są długołańcuchowe kwasy tłuszczowe. Jednak serce ma ogromną elastyczność metaboliczną przejawiającą się zdolnością do utylizowania nie tylko kwasów tłuszczowych, ale także glukozy, mleczanów i ciał ketonowych. Endokannabinoidy mogą bezpośrednio oddziaływać na kardiomiocyty poprzez obecne na ich powierzchni receptory CB1 i CB2. Wydaje się, że bezpośrednia aktywacja receptorów CB1 sprzyja wzmożonej lipogenezie, stłuszczeniu osierdzia i zaburzeniom czynności bioelektrycznej serca. Natomiast pobudzone receptory CB2 wykazują właściwości kardioprotekcyjne, przyczyniając się do zachowania odpowiedniego stężenia ATP w kardiomiocytach. Ponadto, działanie endokannabinoidów zarówno na poziomie ośrodkowego układu nerwowego, jak i tkanek obwodowych, tj. wątroby, trzustki czy też tkanki tłuszczowej, pośrednio przekłada się na dostępność osoczową substratów energetycznych i wpływa na metabolizm mięśnia sercowego. Niewiele jest bezpośrednich dowodów opisujących skutki aktywacji układu endokannabinoidowego w warunkach fizjologicznych w układzie krążenia. W pracy opisano wpływ chorób metabolicznych, m.in. otyłości i cukrzycy, a także chorób układu krążenia – nadciśnienia, niedokrwienia i zawału na deregulację układu endokannabinoidowego z następczym wpływem na metabolizm kardiomiocytów.
Układ endokannabinoidowy
Układ endokannabinoidowy jest endogennym, kompleksowym systemem sygnałowym biorącym udział w licznych procesach fizjologicznych. Układ ten tworzą:
• receptory endokannabinoidowe (CB – cannabinoid receptor) z rodziny receptorów 7-transbłonowych,
• endokannabinoidy, czyli endogenne ligandy receptorów CB,
• enzymy zaangażowane w syntezę, wychwyt i degradację endokannabinoidów [23,43].
Dotychczas najwięcej informacji o układzie endokannabinoidowym pochodzi z badań nad ośrodkowym układem nerwowym [33].
Receptory endokannabinoidowe
Dotąd odkryto dwa główne rodzaje receptorów endokannabinoidowych – receptor typu I – CB1 oraz receptor typu II – CB2 [48,90]. Zarówno receptor CB1 jak i CB2 należą do grupy receptorów metabotropowych sprzężonych z biał- kiem G i wykazują homologię w 44% [48].
Receptor CB1 jest zbudowany z 472 aminokwasów i jest kodowany przez gen CNR1 [13,61]. Receptory CB1 są umiejscowione głównie na zakończeniach presynaptycznych neuronów ośrodkowego i obwodowego układu nerwowego. Ponadto w mózgu wykryto receptor CB1A, który jest białkowym produktem genu kodującego receptor CB1, powstałym w wyniku jego alternatywnego składania [48]. Należy podkreślić, że receptory CB1 poza ośrodkowym układem nerwowym wykazują ekspresję w licznych narządach obwodowych, tj. mięśniu sercowym, mięśniach szkieletowych, płucach, jądrach, nasieniowodach, macicy, pęcherzu moczowym, nabłonku jelita cienkiego i grubego, a także w tkance tłuszczowej [7,31,81]. Z dotychczas przeprowadzonych badań wynika, że aktywacja receptorów CB1 występujących w mięśniu sercowym wywołuje skutek inotropowy ujemny, natomiast aktywacja receptorów CB1 w obrębie naczyń wieńcowych powoduje ich wazodylatację [7,102]. Ponadto ekspresję receptorów CB1 wykryto w hodowlach komórek śródbłonka tętnic wień- cowych, a także w kardiomiocytach [73,88]. Natomiast w badaniach in vivo stwierdzono obecność receptorów CB1 na zakończeniach presynaptycznych włókien współ- czulnych unerwiających naczynia oporowe i serce oraz na zakończeniach postsynaptycznych włókien współczulnych w naczyniach krwionośnych [62,73,88,89,91]. W badaniach przeprowadzonych na szczurach wykazano, że aktywacja receptorów CB1 występujących na zakończeniach presynaptycznych włókien współczulnych naczyń oporowych i wieńcowych hamuje sekrecję noradrenaliny z tych zakończeń, co prowadzi do zmniejszenia neurogennej odpowiedzi presyjnej i tachykardii [62,91].
Receptor CB2 jest utworzony z 360 aminokwasów, a za jego ekspresję odpowiada gen CNR2 [13]. Ekspresję receptora CB2 wykazano głównie na komórkach i narzą- dach wchodzących w skład układu immunologicznego, a także w strukturach ośrodkowego układu nerwowego, w tym komórkach mikrogleju [99]. Stwierdzono również obecność receptorów CB2 w kardiomiocytach, kardiomioblastach, a także w komórkach śródbłonka tętnic wieńcowych i komórkach mięśni gładkich naczyń krwionośnych [73,86,87,94]. Uważa się, że receptory CB2 są zaangażowane głównie w regulację procesów odpowiedzi immunologicznej [82]. Wykazano, że aktywacja receptorów CB2 w komórkach śródbłonka naczyń wieńcowych zmniejsza wytwarzanie cytokin przeciwzapalnych, w tym czynnika martwicy guza α (TNF-α – tumor necrosis factor alfa) [86]. Tym samym, wpływa to na chemotaksję i adhezję komórek układu odpornościowego do zaktywowanego śródbłonka [86]. Natomiast aktywacja receptora CB2 w komórkach mięśni gładkich tętnic wieńcowych powoduje zahamowanie ich proliferacji [87].
Wyniki wielu prac z ostatnich lat wykazują istnienie jeszcze innych rodzajów receptorów regulujących homeostazę ustroju z udziałem układu endokannabinoidowego. W danych literaturowych dość często wspominana jest obecność tzw. hipotetycznego receptora śródbłonkowego [19,51,89]. Potwierdzeniem powyższego stwierdzenia wydaje się występowanie rozkurczu izolowanej tętnicy krezkowej i towarzyszący temu spadek ciśnienia krwi pod wpływem anandamidu zarówno u myszy transgenicznych CB1-/-/CB2-/-, jak i u myszy niemodyfikowanych genetycznie [51]. Ponadto zniszczenie śródbłonka naczyniowego i podanie rimonabantu (antagonisty receptorów CB1) w stężeniu wyższym niż jest to wymagane do zablokowania receptorów CB1 zniosło opisaną wyżej zarówno wazodylatację, jak i hipotensję [41,51].
Ligandy receptorów endokannabinoidowych
Endogenne ligandy receptorów endokannabinoidowych są pochodnymi kwasu arachidonowego, będącego metabolitem fosfolipidów błon komórek nerwowych [23]. Wykazano, że endokannabinoidy nie są magazynowane, a ich synteza odbywa się natychmiast po zadziałaniu bodźca w postaci potencjału czynnościowego, jest to tzw. „synteza na żądanie” [27]. Uwolnienie endokannabinoidów do przestrzeni synaptycznej następuje podczas depolaryzacji błony komórkowej elementu postsynaptycznego neuronu w wyniku następczego napływu jonów wapnia do wnętrza komórki [14]. Stwierdzono też, że endokannabinoidy należą do transmiterów wstecznych, tzn. są uwalniane z zakończenia postsynaptycznego, a wpływają na element presynaptyczny, gdzie są obecne ich receptory [4,28]. Mechanizm komunikacji między neuronami nazwano indukowanym depolaryzacją zmniejszaniem hamowania (DSI – depolarization-induced suppression of inhibition) lub indukowanym depolaryzacją zmniejszaniem pobudzania (DSE – depolarization-induced suppression of excitation). Zależy to od tego, czy zablokowane zostaje wydzielanie neuromediatorów hamujących, np. kwasu γ-aminomasłowego, czy też pobudzających, np. noradrenaliny, czy też serotoniny z elementu presynaptycznego neuronu [75].
Jak dotąd udało się zidentyfikować cztery związki wykazujące działanie agonistyczne w stosunku do receptorów endokannabinoidowych. Pierwszym z odkrytych endokannabinoidów był anandamid (AEA), który wyizolowano z mózgu świni [21]. Wykazuje większe powinowactwo do receptora CB1 (którego jest częściowym agonistą) ani- żeli CB2 [90]. Anandamid pobudza również receptory N- -metylo-D-asparaginowe (receptory NMDA) [48]. Innym z odkrytych agonistów był 2-arachidonyloglicerol (2-AG), który ma mniejsze powinowactwo do receptora CB1 w porównaniu z anandamidem, a jednocześnie jest pełnym agonistą tego receptora, co oznacza, że po związaniu z receptorem CB1 wywołuje większy efekt niż anandamid. Zarówno w badaniach in vivo, jak i in vitro, wykazano, że oba związki charakteryzują się małą stabilnością wynikająca z budowy chemicznej i szybko ulegają hydrolizie [90]. Innym ligandem w układzie endokannabinoidowym jest eter noladyny, mający podobnie jak 2-AG niskie powinowactwo do receptora CB1. Jednak eter noladyny ze względu na obecność wiązania eterowego ma większą stabilność w porównaniu z poprzednimi agonistami, co może wpływać na wydłużenie wywoływanego przez ten związek działania biologicznego [40]. Do grupy agonistów receptorów endokannabinoidowych zalicza się również endovanilloid (zwany też N-arachidonoyletanolaminą, dopaminą N-arachidonylową, NADA), będący agonistą zarówno receptora CB1, jak też receptora waniloidowego typu 1 (TRPV1) [25]. Istnieją także inne związki wykazujące aktywność wobec receptorów endokannabinoidowych, m.in. palmitoiloetanolamid (PEA), oleiloetanolamid (OEA), będące odpowiednio pochodnymi kwasu palmitynowego i kwasu oleinowego [8,54]. Palmitoiloetanolamid może aktywować receptor CB1, a także CB2 [8]. Natomiast oleiloetanolamid wykazuje niewielkie powinowactwo wobec receptora CB1, przy czym w ogóle nie wpływa na receptory CB2 [54,76].
Jedynym zbadanym endogennym ligandem, który wykazuje cechy antagonistyczne wobec receptorów endokannabinoidowych jest wirodhamina [83]. Synteza tego związku jest związana z estryfikacją kwasu arachidonowego przez etanolaminę. Należy podkreślić, że wirodhamina wykazuje właściwości antagonistyczne tylko w stosunku do receptora CB1, podczas gdy wobec receptora CB2 działa agonistycznie [83]. Poza aktywacją receptorów metabotropowych w komórkach docelowych, endokannabinoidy mają zdolność bezpośredniej aktywacji receptorów jonotropowych, m.in. receptorów TRPV1, serotoninowych 5HT3 oraz nikotynowych dla acetylocholiny z podjednostką α7 [79].
Enzymy i białka transportowe biorące udział w metabolizmie endokannabinoidów
Zgodnie z obecną wiedzą, endokannabinoidy po wydzieleniu do przestrzeni synaptycznej ulegają wychwytowi zwrotnemu. Jest to możliwe dzięki obecności białek transportujących endokannabinoidy, znajdujących się w błonie elementu postsynaptycznego. Najlepiej poznano białko swoiste dla anandamidu zaangażowane w jego wychwyt zwrotny i wewnątrzkomórkowy transport, które nazwano FAAH-podobnym transporterem anandamidu (FLAT – FAAH-like anandamide transporter) [34,64]. Wśród innych białek, które najprawdopodobniej mogą być związane z transportem wewnątrzkomórkowym endokannabinoidów wyróżnić należy: białko wiążące kwasy tłuszczowe 5 i 7 (FABP 5 and 7 – fatty acid binding protein 5 and 7), białko szoku cieplnego 70 (HSP 70 – heat shock protein 70) oraz albuminy [5,52,78]. Wychwycone przez białka transportowe endokannabinoidy pod wpływem enzymu hydrolazy amidowej kwasów tłuszczowych (FAAH) oraz lipazy monoacyloglicerolowej (MGL) we wnętrzu elementu postsynaptycznego ulegają następnie hydrolizie [45]. Stwierdzono, że w stosunku do AEA największą swoistość wykazuje FAAH, natomiast wobec 2-AG zarówno FAAH, jak i MGL są tak samo swoiste [12,45].
Po raz pierwszy MGL jako hydrolazę serynową inaktywującą 2-AG opisano na podstawie doświadczenia przeprowadzonego na mózgu szczura [29]. Powstający w wyniku hydrolizy kwas arachidonowy, może się stać substratem do syntezy m.in. prostaglandyn, co wskazuje, iż prawdopodobnie inhibicja MGL niesie za sobą ogromny potencjał przeciwbólowy i przeciwzapalny [75]. W badaniach przeprowadzonych na szczurach, którym indukowano ból stwierdzono, że zablokowanie rozkładu 2-AG przez inhibitory MGL wywołuje efekt antynocyceptywny zależny od aktywacji receptorów CB1 bądź CB2 [39]. Jednocześnie w innych badaniach wykonanych na myszach, z wyindukowanym zapaleniem jelit, dowiedziono, że po zastosowaniu inhibitorów MGL następuje zmniejszenie ekspresji cytokin prozapalnych, tj. IL-1β i TNF-α [1]. Należy podkre- ślić, że obecność enzymu MGL wykazano również w sercu szczura [53]. Mimo braku badań, nie jest wykluczone, że zastosowanie inhibitorów MGL może mieć znaczenie w zmniejszeniu syntezy prostaglandyn i ograniczeniu rozwoju stanu zapalnego podczas epizodów niedokrwienia mięśnia sercowego.
Znacznie więcej doniesień dotyczy roli FAAH w regulacji układu endokannabinoidowego [16,74,80]. Wyróżnia się dwie izoformy enzymu FAAH – FAAH-1 oraz FAAH-2. Enzym FAAH-1 wykazuje ekspresję u wszystkich ssaków, podczas gdy FAAH-2, nie przejawia ekspresji u wszystkich gatunków, m.in. nie występuje u szczurów [25]. Wewnątrzkomórkowa hydrolityczna aktywność tych enzymów pozwala utrzymać dokomórkowy gradient stężeń endokannabinoidów na odpowiednim poziomie [5]. Najprawdopodobniej enzym FAAH wpływa na możliwości modulowania sygnału przekazywanego przez układ endokannabinoidowy, umożliwiając zastosowanie FAAH w terapii bólu, nudności, a także przeciwdziałaniu uszkodzeniom serca powstałym np. na wskutek przyjmowania leków kardiotoksycznych (m.in. doksorubicyny) [74]. Wykazano, że u myszy pozbawionych genu kodującego FAAH (FAAH-/-) wzrosta poziom anandamidu w miokardium w porównaniu do myszy szczepu dzikiego (FAAH+/+) [80]. Ponadto myszy FAAH-/- w porównaniu do myszy nieznokautowanych wykazują większą wrażliwość na dzia- łanie depresyjne egzogennego anandamidu w układzie sercowo-naczyniowym. Potwierdzeniem obserwacji jest wydłużenie czasu hipotensji zależnej od aktywacji receptorów CB1 oraz zmniejszenie kurczliwości mięśnia sercowego w trójfazowej odpowiedzi hemodynamicznej na dożylne podanie anandamidu u myszy FAAH-/- [80]. Prawdopodobnie jest to wynikiem braku szybkiej inaktywacji egzogennie podanego anandamidu przez różne tkanki [16,80], w tym miokardium [80]. Jednocześnie myszy FAAH-/- zachowują prawidłową czynność serca, wartość ciśnienia krwi oraz wrażliwość baroreceptorów mimo wzrostu poziomu endogennego anandamidu w mięśniu sercowym tych zwierząt [80].
Wiedza na temat enzymatycznej regulacji układu endokannabinoidowego ulega stałemu pogłębianiu. Istnieją doniesienia na temat licznych enzymów, m.in. lipooksygenazy, cytochromu P450, cyklooksygenazy 2, które mogą przekształcać endokannabinoidy do bioaktywnych produktów (pochodnych eikozanoidów, np. etanolamidu prostaglandyny E2), których fizjologiczna rola pozostaje jak dotąd niewyjaśniona [17,103]. Zmiany stężeń zarówno transporterów jak i enzymów mogą zaburzać metabolizm i aktywność endokannabinoidów wytwarzanych w tkankach [45].
Mechanizm aktywacji postreceptorowej w układzie endokannabinoidowym
Pobudzenie receptorów CB1 i CB2 przez swoiste ligandy powoduje aktywację białka Gi/o [48], co w obu przypadkach wywołuje spadek aktywności cyklazy adenylowej i wewnątrzkomórkowego stężenia cAMP [99]. W przypadku receptorów CB1 sugeruje się, że aktywacja białka Gi/o pobudza kaskady kinaz aktywowanych mitogenami (MAPK), odbywającej się przez aktywację kinazy 3-fosfatyloinozytolu [46,48]. Receptory CB1 poprzez aktywację białka Gi/o wpływają na regulację czynności błonowych kanałów jonowych wapnia (kanały typu N, L, P/Q) i potasu (kanałyypu A i Ir). W wyniku aktywacji receptorów CB1 obecne w błonie komórkowej komórek docelowych kanały wapniowe ulegają zamknięciu, przy czym nastę- puje aktywacja i otwarcie kanałów potasowych [48]. Aktualny stan wiedzy wskazuje, że w przypadku aktywacji receptorów CB2 w szlaku transmisji sygnału bierze udział kinaza białkowa C [46] i jak dotąd nie stwierdzono, aby aktywacja receptorów CB2 wpływała na działanie kana- łów jonowych [98].
Ponadto obecne w błonie komórkowej receptory CB1 są związane ze specjalnymi mikrodomenami błonowymi, zwanymi „tratwami lipidowymi” (LR – lipid rafts), które zawierają duże stężenia cholesterolu i glikosfingolipidów. Tratwy lipidowe modulują zdolność wiązania ligandów z receptorami CB1 i wpływają na wewnątrzkomórkowe CB1-zależne szlaki sygnalizacyjne [3]. W badaniach na hodowlach C6 komórek glejaka stwierdzono, iż błonowa zawartość cholesterolu wpływa na integralność tratw lipidowych z błoną komórkową i moduluje tym samym interakcję endokannabinoidów z receptorami [3]. W tych badaniach, w wyniku usuwania cholesterolu z błony komórkowej przez metylo-β-cyklodekstrynę, zauważono zależne od dawki metylo-β-cyklodekstryny zwiększenie wiązania receptorów CB1 ze swoistymi ligandami, m.in. [3H]CP55.940 [3]. Ponadto nastąpił też wzrost przekazywania sygnału zależnego od białek Gi/o związany ze zwiększeniem aktywacji szlaków sygnałowych cyklazy adenylowej i MAPK [3].
Specyfika metabolizmu mięśnia sercowego
W warunkach fizjologicznych w mięśniu sercowym zachodzi metabolizm tlenowy, którego zadaniem jest ciągła resynteza ATP. W celu pozyskania energii są wykorzystywane w 60-70% długołańcuchowe kwasy tłuszczowe (LCFA), w 20-30%, glukoza, a także mleczany i ciała ketonowe [36,96]. Wykazano, że wybór substratu energetycznego zależy od jego dostępności we krwi krążącej, a także od zmian zapotrzebowania energetycznego mię- śnia sercowego [96].
Ze względu na to, iż LCFA stanowią jeden z podstawowych substratów energetycznych mięśnia sercowego, przeprowadzono wiele badań, które dostarczyły istotnych informacji o ich dokomórkowym transporcie. Wykazano, że LCFA dostają się do wnętrza kardiomiocytów na zasadzie biernej dyfuzji (20%) [6] lub z udziałem swoistych białek transportujących kwasy tłuszczowe w procesie transportu wspomaganego (80%) [100]. Do białek tych w mięśniu sercowym zalicza się: FAT/CD36 – translokazę kwasów tłuszczowych, FABPpm – błonowe białko wiążące kwasy tłuszczowe oraz FATP-1, FATP-4 i FATP-6 – białka transportujące kwasy tłuszczowe typu 1, 4 i 6 [30,36]. Ustalono, że białka te znajdują się nie tylko w błonie kardiomiocytów, ale również tworzą pulę zgromadzoną w pęcherzykach wewnątrzkomórkowych mającą zdolność translokacji na powierzchnię błony komórkowej [96]. Translokacja białek transportujących LCFA do błony komórkowej, m.in. pod wpływem czynności skurczowej kardiomiocytów [92], czy też w wyniku stymulacji hormonalnej, np. insuliną [59] zwiększa napływ LCFA do wnętrza kardiomiocytów [59]. W ten sposób możliwe jest dostosowanie podaży substratów energetycznych do aktualnego zapotrzebowania miokardium na energię [6,59]. LCFA po wejściu do wnętrza kardiomiocytów są wiązane przez cytoplazmatyczne białko wiążące kwasy tłuszczowe typu sercowego (H-FABPc) [6]. W takiej postaci LCFA mogą podlegać aktywacji enzymatycznej przez syntetazę acylo-CoA (FACS) po związaniu z koenzymem A (CoA). Aktywny kompleks enzym-substrat łączy się z białkami wiążącymi acylo-CoA (ACBP), co powoduje przemieszczenie LCFA-CoA na dalszy tor przemian w kierunku β-oksydacji lub estryfikacji do określonych frakcji lipidowych [96]. W pierwszym przypadku LCFA związane z CoA dostają się do wnętrza mitochondrium z udziałem enzymu umiejscowionego na zewnętrznej błonie mitochondrialnej – palmitoilotransferazie karnitynowej I (CPT I) oraz karnitynie [58]. Dochodzi do wytworzenia acylokarnityny i uwolnienia CoA. Transport między błoną zewnętrzną a wewnętrzną mitochondrium odbywa się z udziałem kolejnego enzymu, którym jest acylotranslokaza karnitynowa (CAT). Aby doszło do utleniania kwasów tłuszczowych reszta acylowa musi ulec odłączeniu od karnityny i połączyć się z CoA znajdującym się wewnątrz mitochondrium. Na tym etapie katalizowana jest palmitoilotransferaza karnitynowa II. Gdy zostaje odtworzony acylo-CoA, w macierzy mitochondrialnej następuje utlenianie kwasów tłuszczowych, mające na celu wytworzenie energii w postaci ATP, odbywające się dwuetapowo. Pierwszym etapem jest β-oksydacja, podczas której zostaje wytworzony aktywny octan, a następnie jest włączany w cykl kwasów trójkarboksylowych (etap drugi) z wytworzeniem produktów końcowych – dwutlenku węgla oraz wody [18,58]. Zaktywowane LCFA w mniejszym stopniu (10-30%) mogą również tworzyć wewnątrzkomórkową pulę kwasów tłuszczowych z udziałem acylotransferazy glicerolofosforanowej, tworząc wewnątrzkomórkowe zasoby mono-, di- i triacylogliceroli, uwalnianianych w razie zaistnienia zapotrzebowania na LCFA [96].
Jak już wspomniano, drugim pod względem wielkości substratem energetycznym wykorzystywanym przez mię- sień sercowy jest glukoza. Wykazano, że transport glukozy do wnętrza kardiomiocytów odbywa się z udziałem błonowych transporterów GLUT. W metabolizm glukozy w mięśniu sercowym są zaangażowane transportery GLUT1 (przeważając w okresie płodowym) i GLUT4 [18,32]. Transportery GLUT1 są umiejscowione głównie w błonie komórkowej i nie wykazują wrażliwości na stymulację insuliną [32]. Natomiast transportery GLUT4 znajdują się głównie wewnątrz komórki w pęcherzykach mikrosomalnych. Translokacja GLUT4 na powierzchnię błony komórkowej zachodzi podczas stymulacji przez róż- ne czynniki, wśród których wymienić należy insulinę, niedokrwienie oraz przeciążenie serca (overload) [32]. W warunkach fizjologicznych w sercu występuje również niska ekspresja transportera GLUT3 [20]. Oprócz wymienionego transportu istnieje również kotransport sodu i glukozy z udziałem transportera SGLT1, reagującego na stymulację insuliną oraz leptyną [2]. Glukoza po wejściu do wnętrza komórki w wyniku procesu glikolizy przekształcana zostaje w pirogronian. W warunkach tlenowych pirogronian pod wpływem enzymu dehydrogenazy pirogronianowej (PDH) przekształca się do acetylo-CoA, który jest włączany do cyklu kwasów trójkarboksylowych (Krebsa). Przy braku dostępności tlenu dehydrogenaza pirogronianowa (LDH) powoduje powstawanie mleczanu z pirogronianu, jednak należy podkreślić, iż procesy glikolizy beztlenowej w mięśniu sercowym są znikome [18].
Wpływ endokannabinoidów na metabolizm mięśnia sercowego
Pobór substratów energetycznych przez mięsień sercowy zależy od wielu czynników, wśród których wymienić należy: stężenie docierających do kardiomiocytów substratów, wpływ hormonów regulujących metabolizm (m.in. insulina), zapotrzebowanie serca na energię (wysiłek fizyczny, tachykardia, wzrost obciążenia następczego) oraz zaopatrzenie miokardium w tlen [96]. Pośrednio lub bezpośrednio każdy czynnik może aktywować układ endokannabinoidowy. Brak jest danych literaturowych opisujących bezpośredni wpływ endokannabinoidów na metabolizm miokardium. Ponieważ receptory tego układu są umiejscowione nie tylko na powierzchni kardiomiocytów, ale również występują na zakończeniach włókien współczulnych autonomicznego układu nerwowego unerwiającego serce, z tego można wnioskować, iż endokannabinoidy są zaangażowane w metabolizm mięśnia sercowego w sposób bezpośredni. Możliwe jest również, że endokannabinoidy wykazują wpływ pośredni, modulując transmisję neuronalną przez umiejscowione w ośrodkowym i obwodowym układzie nerwowym komponenty tego układu, wpływając na podwzgórzową regulację poboru pokarmu, metabolizm substratów energetycznych w wątrobie i ostatecznie zmieniając przepływ wieńcowy [15,48].
Pośredni wpływ endokannabinoidów na metabolizm mięśnia sercowego
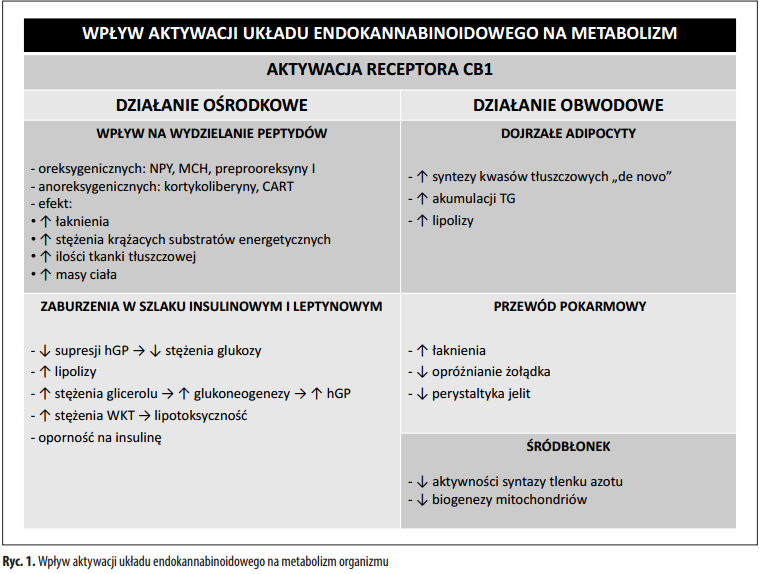
Wiele danych doświadczalnych dowodzi wpływu układu endokannabinoidowego na wzmożone łaknienie [9,15,35,49]. Działanie to jest możliwe przez wpływ na receptory CB1 obecne w obrębie podwzgórza (jądro przykomorowe i jądro łukowate podwzgórza). W badaniach przeprowadzonych na szczurach udowodniono, że przez bezpośrednią iniekcję anandamidu do podwgórza można wywołać uczucie głodu [49]. Ponadto endokannabinoidy aktywując receptory CB1 w podwgórzu działają na uwalnianie peptydów oreksygenicznych regulujących pobieranie pokarmu, np. neuropeptyd Y, hormon melanocytotropowy czy preprooreksynę 1. Ekspresję receptorów CB1 stwierdzono również w neuronach podwzgórza wydzielających peptydy anoreksygeniczne, czyli zmniejszające łaknienie, m.in. kortykoliberynę, transkrypt regulowany przez kokainę i amfetaminę. Prawdopodobnie przez aktywację receptorów CB1 jest możliwe wpływanie na ich wydzielanie i tym samym regulację apetytu [15,35,49].
Aktywacja receptorów CB1 w obrębie układu mezolimbicznego reguluje nie tylko łaknienie, ale bierze też udział w procesach motywacji i aktywacji behawioralnej w wyniku zadziałania czynnika nagradzającego (konsumpcji pokarmu) [9,15]. W badaniu przeprowadzonym na zdrowych ochotnikach wykazano, że konsumpcji ulubionych potraw w porównaniu do potraw o neutralnych walorach smakowych towarzyszy wzrost stężenia 2-arachidonyloglicerolu w osoczu, który pozytywnie koreluje z osoczowym stężeniem greliny [71]. Wynik eksperymentu może świadczyć o prawdopodobnym zaangażowaniu aktywacji układu endokannabinoidowego w występowanie hiperfagii hedonicznej, czyli spożywania pokarmów dla przyjemności, a nie w celu utrzymania homeostazy energetycznej [71]. Nadmierna podaż pokarmu przyczynia się do wzrostu dostępności substratów energetycznych w osoczu, co istotnie wpływa na energetykę mięśnia sercowego, ponieważ duże stężenie krążących kwasów tłuszczowych bezpośrednio wzmaga dokomórkowy napływ LCFA do kardiomiocytów. Mimo iż kwasy tłuszczowe są podstawowym źródłem energii dla miokardium to wzrost ich metabolizowania wywiera negatywne skutki. Dzieje się tak dlatego, że wraz ze wzrostem oksydacji kwasów tłuszczowych jako substratu energetycznego maleje udział glukozy (a także mleczanów) w przemianach metabolicznych kardiomiocytów. Powyższa zmiana w metabolizmie miokardium zmniejsza wydolność mechaniczną serca, a także zwiększa ilość zu- żywanego tlenu do wygenerowania ATP niezbędnego do skurczu [96]. Ponadto w doświadczeniu przeprowadzonym na myszach wykazano, że nawet podczas 3-dniowego zastosowania diety bogatej w tłuszcze dochodzi do przejściowego wzrostu poziomu 2-arachidonyloglicerolu w podwzgórzu [44]. Natomiast w przypadku wydłużenia okresu stosowania tej diety do 14 dni, stwierdzono trwały wzrost 2-arachidonyloglicerolu w podwzgórzu [44]. W obydwu opisanych doświadczeniach zauważono zwiększenie preferencji do spożywania pokarmów obfitujących w tłuszcze, ustępujące po zastosowaniu antagonisty receptora CB1. Informacje te wskazują, że w wyniku nadmiernej aktywacji receptorów CB1 pod wpływem podwyższonego stężenia 2-arachidonyloglicerolu może wystąpić skłonność do konsumpcji pokarmów wysokotłuszczowych [44]. W wyniku spożywania zwiększonej ilości tłuszczów w diecie wzrasta stężenie krążących wolnych kwasów tłuszczowych, co w rezultacie wpłynie na zwiększenie dostępności tego substratu energetycznego dla kardiomiocytów. Di Marzo i wsp. [26] w badaniach na zwierzętach wykazali ponadto, że w podwzgórzu występuje pewien rodzaj sprzężenia zwrotnego między 2-AG a leptyną. Zauważono bowiem, że podwyższonemu stężeniu 2-AG towarzyszy upośledzenie sygnału przekazywanego przez leptynę u otyłych myszy db/db i ob/ob i szczurów Zucker [26]. Natomiast jednorazowe podanie leptyny redukuje stężenie 2-AG u myszy ob/ob i szczurów bez mutacji genetycznej [26]. Powyż- sze odkrycie wskazuje na udział 2-AG w podwzgórzu jako komponenty wchodzącej w skład obwodów neuronalnych regulowanych przez leptynę. Ponadto toniczne pobudzanie receptorów CB1 przez 2-AG najprawdopodobniej ma na celu podtrzymanie pobierania pokarmu [26]. Sugeruje się, że podwyższone stężenie 2-AG i towarzyszące mu zaburzenia w szlaku leptynowym [26] mogą mieć udział w obwodowych zaburzeniach metabolicznych, np. w wą- trobowej oporności na insulinę [77]. Wykazano, że aktywacja receptorów CB1 w obrębie ośrodkowego układu nerwowego u szczurów z infuzją agonistów receptorów CB1 – WIN55,212-2 (WIN) lub arachidonoilo-2-chloroetylamidu (ACEA) do komór mózgu hamuje zależną od insuliny supresję wątrobowego wytwarzania glukozy, co powoduje nadmierne wytwarzanie glukozy przez wątrobę [77]. Zaobserwowane zahamowanie supresji wątrobowej wytwarzanej glukozy nie wynika ze zmian w wewnątrzkomórkowym szlaku insulinowym w wątrobie, ponieważ zarówno ekspresja, jak i fosforylacja istotnych składowych tego szlaku – kinazy białkowej AKT oraz kinazy syntazy glikogenu 3 – GSK3 w wątrobie nie zmieniły się pod wpływem WIN i ACEA [77]. Najprawdopodobniej zahamowanie zależnego od insuliny, wątrobowego wytwarzania glukozy pod wpływem WIN i ACEA jest wynikiem zaburzeń w szlaku insulinowym i leptynowym na poziomie ośrodkowego układu nerwowego, przy czym mechanizm ten nie został wyjaśniony [56]. Ponadto w wyniku centralnej aktywacji receptorów CB1 upośledzona zostaje zdolność insuliny do hamowania ogólnoustrojowej lipolizy białej tkanki tłuszczowej (WAT – white adipose tissue) wskutek zwiększenia ekspresji i fosforylacji białka przenoszącego lipidy – perilipiny oraz koaktywatora lipazy triacyloglicerolowej (ATGL – adipose triglyceride lipase) – CGI-58 (comparative gene identification-58) [77]. Dochodzi także do wzrostu ekspresji głównych enzymów lipolitycznych w WAT – ATGL i lipazy hormonowrażliwej (HSL – hormone-sensitive lipase). Uwolniony z WAT glicerol może stanowić podłoże do glukoneogenezy w wątrobie, zwiększając tym samym stężenie glukozy we krwi i mechanizm obserwowanej w wątrobie oporności na insulinę [77]. Zwiększona lipoliza przyczynia się do podwyższenia stężenia krążących wolnych kwasów tłuszczowych, a przez to do wygenerowania lipotoksyczności, która jest jednym z czynników patogenetycznych w rozwoju cukrzycy typu 2. Powyższe zmiany wpływają na zwiększenie dostępności substratów energetycznych dla wszystkich komórek, w tym również dla kardiomiocytów. Zwiększenie osoczowej biodostępności glukozy i kwasów tłuszczowych zwiększa dokomórkowy transport tych substratów energetycznych do wnętrza kardiomiocytów [26,77,93].
Ze względu na obecność receptorów endokannabinoidowych w obrębie tkanek i narządów obwodowych o znacznej aktywności metabolicznej, endokannabinoidy wpływają na gospodarkę energetyczną ustroju także poza ośrodkowym układem nerwowym. Wykazano, że u myszy szczepu dzikiego obecne w dojrzałych adipocytach receptory CB1 w wyniku aktywacji w warunkach in vitro powodują zwiększenie syntezy kwasów tłuszczowych de novo, wewnątrzkomórkową akumulację triglicerydów oraz zmniejszenie lipolizy [101]. Podłożem zmian jest pobudzenie wychwytu glukozy i aktywacja enzymu lipogenezy, tj. syntazy kwasów tłuszczowych (FAS – fatty acid synthase). Prawdopodobnie prowadzi to do zmniejszenia stężenia wolnych kwasów tłuszczowych i glukozy we krwi. Ponadto, w wyniku aktywacji receptora CB1 dochodzi do zahamowania śródbłonkowej syntazy tlenku azotu i następczego spadku biogenezy i aktywności enzymów mitochondrialnych w adipocytach [101].
Stwierdzono, że receptory CB1 znajdujące się na zakończeniach nerwowych przewodu pokarmowego pod wpływem anandamidu stymulują łaknienie, wpływając tym samym na zwiększenie pobierania pokarmu i wzrost dostępnych w ustroju substratów energetycznych [15]. Przypuszcza się, że endokannabinoidy hamują opróż- nianie żołądka oraz zmniejszają perystaltykę jelit, co również sprzyja zwiększonemu wchłanianiu substratów energetycznych do krwiobiegu. Obecność receptorów CB1 w wątrobie, mimo iż występują w niewielkiej ilości, wpływa na możliwość regulowania metabolizmu glukozy przez endokannabinoidy. Wykazano, że systemowa blokada receptorów CB1 przez antagonistę AVE1625 u szczurów wywołuje wzrost wątrobowej glikogenolizy, co świadczy o prawdopodobnym udziale aktywacji receptorów CB1 w tworzeniu glikogenu w wątrobie [42]. Być może centralna aktywacja receptorów CB1 ma na celu dostarczenie substratów energetycznych dla organizmu, natomiast aktywacja obwodowych receptorów CB1 sprzyja efektywnemu gromadzeniu pozyskanych źródeł energii [24].
Przez obecność receptorów CB1 i CB2 w komórkach β trzustki endokannabinoidy mogą wpływać na metabolizm regulując wydzielanie insuliny. Nie jest jednak pewne, czy aktywacja receptorów CB1 kontroluje tylko sygnał przekazywany przez insulinę czy wpływa na jej wydzielanie [95]. Liu i wsp. [56] w badaniach przeprowadzonych na ludzkich i mysich hepatocytach sugerują, że aktywacja receptorów CB1 pod wpływem anandamidu powoduje zahamowanie sygnału przekazywanego przez insulinę w dwojaki sposób. Indukuje fosforylację seryny w pozycji 307 substratu receptora insuliny typu 1 (IRS1 – insulin receptor substrate 1), co wpływa na zahamowanie aktywności kinazy białkowej B (AKT2). Aktywacja receptorów CB1 wpływa bezpośrednio na zahamowanie indukowanej insuliną fosforylacji AKT2. Kinaza AKT2 będąc w postaci nieufosforylowanej jest nieaktywna i tym samym hamowany jest szlak insulinowy [56]. Główną funkcją AKT2 jest aktywacja kaskady reakcji prowadzących do zwiększenia translokacji transporterów GLUT4 z puli wewnątrzkomórkowej do błony komórkowej pod wpływem stymulacji insuliną [38]. Oznacza to, że glukoza mimo obecności insuliny nie może zostać przetransportowana do wnętrza hepatocytów [56]. Esposito i wsp. [31] w badaniu przeprowadzonym na hodowli C6 komórek mięśni szkieletowych myszy również zauważyli, że układ endokannabinoidowy wpływa ujemnie na regulację szlaków insulinowych P13K-AKT, a także P13K-PDK-PKCξ, których udział jest niezbędny w wychwycie glukozy z udziałem transporterów GLUT4 [95]. Dowiedziono, że komórki α wysp trzustki wytwarzają enzymy biorące udział w syntezie endokannabinoidów, natomiast komórki β syntetyzują enzymy degradujące endokannabinoidy [97]. Zanim endokannabinoidy ulegną hydrolizie, mogą zaktywować receptory CB2 występujące w komórkach β trzustki i spowodować zahamowanie wydzielania insuliny [97]. Ponadto anandamid działając na receptory waniloidowe TRPV1, które są sprzężone z komórkami β trzustki, również wpływa na ich czynność wydzielniczą [97]. Endokannabinoidy przez zahamowanie szlaków insulinowych, a także translokacji transporterów glukozy prawdopodobnie mogą wpłynąć na zmniejszenie stężenia glukozy wewnątrz kardiomiocytów, a tym samym spadek metabolizmu tego substratu energetycznego w miokardium.
Podsumowując wydaje się, że wpływ aktywacji układu endokanabinoidowego na metabolizm jest zarówno ośrodkowy (ogólnoustrojowy) jak i miejscowy (narządowy) (ryc. 1).
Endokannabinoidy w otyłości
Obecnie wiele badań wskazuje, że rozwojowi otyłości towarzyszy aktywacja układu endokannabinoidowego [11,24,37,57,65,66]. Świadczy o tym m.in. występująca u myszy dysregulacja układu endokannabinoidowego powstała w wyniku otyłości wyindukowanej dietą wysokotłuszczową, która objawia się wczesnym i trwałym wzrostem ekspresji anandamidu i 2-AG w wielu tkankach obwodowych, m.in. w sercu [67]. Ze względu na ich właściwości lipogeniczne i jednoczesny wzrost ekspresji receptorów CB1 w sercu może dojść do wzmożenia lipogenezy [67]. Nadmierna lipogeneza może się przyczynić do stłuszczenia osierdzia, co stanowi jeden z czynników ryzyka chorób sercowo-naczyniowych [24,67]. Przeprowadzono także badania mające na celu stwierdzenie czy możliwa jest supresja stymulowanej przez układ endokannabinoidowy lipogenezy. W badaniach tych wykazano, że długotrwałe zablokowanie receptorów CB1 w WAT przez rimonabant zwiększa utlenianie kwasów tłuszczowych i wydatek energetyczny u myszy z otyłością wywołaną dietą wysokotłuszczową [50]. Prawdopodobnie rimonabant wpływa na zwiększenie ekspresji enzymów w WAT zaangażowanych w utlenianie kwasów tłuszczowych [50]. Przeprowadzono też eksperyment na genetycznym modelu otyłości – u szczurów Zucker, któ- rym także podawano rimonabant [69]. Inhibicja receptorów CB1 przyczyniła się do spadku masy ciała, hamowania procesu akumulacji tkanki tłuszczowej, wzrostu wrażliwości na insulinę, zwiększonej aktywności układu współczulnego, a także wzrostu stężenia adiponektyny oraz zmniejszenia ciśnienia krwi [57,69]. Z powyższych doświadczeń wynika, że skutki nadmiernej aktywacji układu endokannabinoidowego mogą być łagodzone przez antagonistę receptorów CB1 zarówno w otyłości wynikającej z nieprawidłowej diety, jak i otyłości spowodowanej czynnikami genetycznymi.
Niemniej interesującym wydaje się badanie, w którym wykazano, że aktywność enzymu FAAH w dojrzałych adipocytach pozytywnie koreluje ze wzrostem wskaźnika masy ciała (BMI – body mass indeks) i obwodem talii u zdrowych osób [11]. Prawdopodobnie wzrost aktywności enzymu FAAH jest wynikiem ogólnego wzrostu aktywności układu endokanabinoidowego w czasie rozwoju otyłości. Jednocześnie obserwacja potwierdza hipotezę, że niektó- re składowe układu endokannabinoidowego są pobudzane w wyniku pojawienia się chorób, m.in. otyłości [11].
Endokannabinoidy w cukrzycy
Podobnie jak w otyłości, istnieje wiele badań wskazują- cych, że aktywacja receptorów CB1 sprzyja rozwojowi cukrzycy typu 2 [57]. Liu i wsp. w badaniach na myszach zauważyli, że po zastosowaniu ich blokera – rimonabantu nastąpiło zwiększenie wychwytu i metabolizmu glukozy, m.in. w mięśniach szkieletowych, co jednocześnie wskazuje na zależne od aktywacji receptorów CB1 zaburzenie przemian metabolicznych glukozy [57]. W celu wyjaśnienia molekularnego mechanizmu zaburzeń metabolizmu glukozy warto jest zwrócić uwagę na wyniki doświadczenia przeprowadzonego przez Motaghediego i wsp. [72]. Wykazali związek między zastosowaniem antagonisty receptorów CB1 a zwiększeniem translokacji transportera glukozy GLUT4 z kompartmentu wewnątrzkomórkowego do błony komórkowej w adipocytach [72]. Oznacza to, że aktywacja receptorów CB1 hamuje translokację transportera GLUT4, a dane literaturowe wskazują, że opisany mechanizm występuje również m.in. w komórkach mięśni szkieletowych [47], co nie wyklucza udziału hamowania translokacji GLUT4 przez zaktywowane receptory CB1 w kardiomiocytach. W wyniku spadku liczby błonowych transporterów glukozy wychwyt glukozy z krwi do komó- rek ulega zmniejszeniu, prowadząc do spadku intensywności metabolizowania glukozy w komórkach. Ponadto badaniom poddano wpływ układu endokannabinoidowego w rozwoju cukrzycy typu 1 [85]. Rajesh i wsp. dowiedli, że cukrzyca typu 1 doprowadza do wzrostu ekspresji receptorów CB1 i wzrostu stężenia anandamidu w sercu myszy [85]. Ponadto dysfunkcja mięśnia sercowego powstająca w przebiegu cukrzycy ulega poprawie u myszy CB1-/- oraz u myszy nieznokautowanych leczonych antagonistami receptorów CB1 [85]. Genetyczna delecja lub farmakologiczna inhibicja receptorów CB1 osłabia aktywację MAPK, zmniejsza apoptozę komórek, stan zapalny oraz stres oksydacyjny i nitrozacyjny obejmujący miokardium myszy z cukrzycą [85]. Wyniki badań otrzymanych Podobnie jak w otyłości, istnieje wiele badań wskazują- cych, że aktywacja receptorów CB1 sprzyja rozwojowi cukrzycy typu 2 [57]. Liu i wsp. w badaniach na myszach zauważyli, że po zastosowaniu ich blokera – rimonabantu nastąpiło zwiększenie wychwytu i metabolizmu glukozy, m.in. w mięśniach szkieletowych, co jednocześnie wskazuje na zależne od aktywacji receptorów CB1 zaburzenie przemian metabolicznych glukozy [57]. W celu wyjaśnienia molekularnego mechanizmu zaburzeń metabolizmu glukozy warto jest zwrócić uwagę na wyniki doświadczenia przeprowadzonego przez Motaghediego i wsp. [72]. Wykazali związek między zastosowaniem antagonisty receptorów CB1 a zwiększeniem translokacji transportera glukozy GLUT4 z kompartmentu wewnątrzkomórkowego do błony komórkowej w adipocytach [72]. Oznacza to, że aktywacja receptorów CB1 hamuje translokację transportera GLUT4, a dane literaturowe wskazują, że opisany mechanizm występuje również m.in. w komórkach mięśni szkieletowych [47], co nie wyklucza udziału hamowania translokacji GLUT4 przez zaktywowane receptory CB1 w kardiomiocytach. W wyniku spadku liczby błonowych transporterów glukozy wychwyt glukozy z krwi do komó- rek ulega zmniejszeniu, prowadząc do spadku intensywności metabolizowania glukozy w komórkach. Ponadto badaniom poddano wpływ układu endokannabinoidowego w rozwoju cukrzycy typu 1 [85]. Rajesh i wsp. dowiedli, że cukrzyca typu 1 doprowadza do wzrostu ekspresji receptorów CB1 i wzrostu stężenia anandamidu w sercu myszy [85]. Ponadto dysfunkcja mięśnia sercowego powstająca w przebiegu cukrzycy ulega poprawie u myszy CB1-/- oraz u myszy nieznokautowanych leczonych antagonistami receptorów CB1 [85]. Genetyczna delecja lub farmakologiczna inhibicja receptorów CB1 osłabia aktywację MAPK, zmniejsza apoptozę komórek, stan zapalny oraz stres oksydacyjny i nitrozacyjny obejmujący miokardium myszy z cukrzycą [85]. Wyniki badań otrzymanych przez Rejesha i wsp. dotyczące wzrostu ekspresji receptorów CB1 i stężenia anandamidu w lewej komorze serca myszy z cukrzycą typu 2 są zgodne z wynikami wzrostu stężenia anandamidu w ludzkiej siatkówce oka objętej retinopatią cukrzycową [68,85]. Powyższe wyniki dowodzą ogólnoustrojowego wpływu układu endokannabinoidowego na organizm podczas cukrzycy.
Endokannabinoidy a układ krążenia
Endokannabinoidy in vivo mają bezpośredni wpływ na układ sercowo-naczyniowy [17]. Badania przeprowadzone na szczurach dowiodły, że anandamid może wywoływać zmiany hemodynamiczne występujące w postaci trzech faz [63]. W fazie I dochodzi do jednoczesnej aktywacji receptorów TRPV1 i wystąpienia bradykardii oraz hipotensji. W fazie II następuje krótkotrwała odpowiedź presyjna. Sugeruje się, że faza ta ma złożony mechanizm, w którym są angażowane struktury ośrodkowe (receptory ß-adrenergiczne i NMDA) i obwodowe (naczynia krwionośne; mechanizm zależny od kanałów wapniowych). Prawdopodobnie w tej fazie dochodzi również do aktywacji receptorów TRPV1. W fazie III obserwuje się wydłużoną hipotensję, w której udział biorą receptory CB1 znajdujące się w naczyniach krwionośnych oraz w sercu. Ich aktywacja prowadzi do zahamowania zarówno odpowiedzi presyjnej, jak i tachykardii [63]. Ponadto wykazano wpływ metabolitu anandamidu powstałego w wyniku hydrolizy pod wpływem enzymu FAAH na kanały potasu zależne od ATP (KATP) w izolowanych szczurzych kardiomiocytach komór serca [55]. Powstały podczas hydrolizy anandamidu kwas arachidonowy wpływa na aktywację kana- łów KATP w szczurzych kardiomiocytach [55]. Kanały te są odpowiedzialne za metabolizm energetyczny komórek, tzn. reagują na zmiany wewnątrzkomórkowego stosunku ATP/ADP i są zamykane gdy stosunek ten wzrasta. Ich otwarcie następuje pod wpływem obniżenia wewnątrzkomórkowej zawartości ATP oraz spadku ekspresji kana- łów potasu zależnych od acetylocholiny (KAch). Otwarcie kanałów potasowych powoduje wypływ jonów potasu z komórki i następczą repolaryzację błony komórkowej kardiomiocytów. Aktywacja kanałów KATP przez metabolit anandamidu ma znaczenie antyarytmiczne i kardioprotekcyjne podczas niedotlenienia mięśnia sercowego [55]. Otwarcie kanałów KATP skraca czas trwania potencja- łu czynnościowego kardiomiocytów i zmniejsza dopływ wapnia przez kanały typu L, co zapobiega nadmiernemu wzrostowi stężenia wapnia w sercu i utrzymuje ilość ATP w kardiomiocytach na odpowiednim poziomie [55]. Układ endokannabinoidowy nie wykazuje tonicznej aktywności w warunkach fizjologicznych w obrębie układu krążenia. Zauważono natomiast, że w procesach patologicznych kompensuje zmiany w parametrach krążenia, m.in. przez zmiany w wydzielaniu endokannabinoidów i ekspresji receptorów endokannabinoidowych [10,62,63,80].
Endokannabinoidy w nadciśnieniu
Wyniki badań wskazują na wpływ aktywacji receptorów CB1 przez endokannabinoidy na obniżenie ciśnienia tętniczego krwi w nadciśnieniu tętniczym u szczurów [10]. W różnych postaciach nadciśnienia tętniczego (samoistnego, nerkopochodnego) występuje toniczna aktywacja układu endokannabinoidowego za co prawdopodobnie odpowiada zwiększona ekspresja receptorów CB1 w sercu i komórkach endotelialnych naczyń krwionośnych [10]. W doświadczeniu przeprowadzonym na szczurach z nadciśnieniem samoistnym (SHR – spontaneously hypertensive rat) stwierdzono, że zwiększona ekspresja receptorów CB1 w kardiomiocytach i komórkach endotelialnych aorty szczurów SHR w porównaniu do szczurów normotensyjnych przekłada się na wzrost działania hipotensyjnego w odpowiedzi na podanie kannabinoidów pochodzenia egzogennego u szczurów SHR [10]. Ponadto zwiększenie aktywacji receptorów CB1 przez zablokowanie degradacji i wewnątrzkomórkowego wychwytu zwrotnego anandamidu normalizuje ciśnienie krwi, o czym świadczy wpływ inhibicji enzymu FAAH przez URB597 u szczurów SHR na zmniejszenie ciśnienia krwi oraz czynności serca do poziomu obserwowanego u zwierząt normotensyjnych. Dodatkowych informacji na temat zależności występujących między nadciśnieniem a układem endokannabinoidowym dostarczyło zastosowanie antagonisty receptorów CB1 – SR141716 u szczurów SHR. U szczurów z nadciśnieniem samoistnym zastosowanie SR141716 wywołuje trwały dalszy wzrost ciśnienia krwi, co sugeruje że prawdopodobnie podniesione podstawowe ciśnienie krwi wpływa na toniczną aktywację receptorów CB1 [10]. Ponadto antagoniści receptorów CB1 zwiększają wydolność skurczową serca bez znaczącego wpływu na obwodowy opór naczyniowy, co może oznaczać, że endokannabinoidy podczas nadciśnienia działają głównie na serce [10]. Antagoniści receptora CB1 oraz inhibitory FAAH zastosowane u szczurów normotensyjnych nie wywoływały żadnego działania. Wyniki tych badań mogą świadczyć o widocznej aktywacji układu endokannabinoidowego w układzie krążenia w stanie hipertensji, a zastosowanie agonistów receptorów CB1 i inhibitorów agonistów receptorów CB1 przyczynia się do normalizacji ciśnienia tętniczego krwi [10].
Endokannabinoidy w niedokrwieniu i zawale mięśnia sercowego
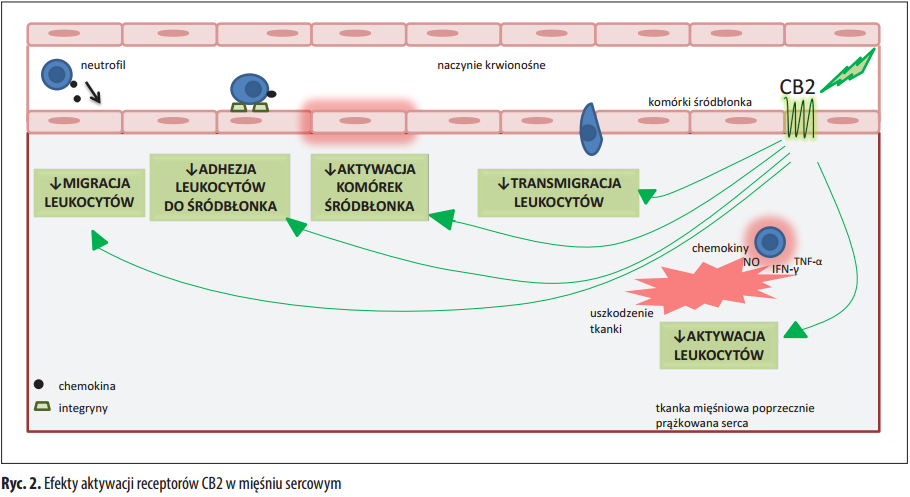
Z dotychczas przeprowadzonych badań wynika, że endokannabinoidy wykazują właściwości kardioprotekcyjne. W eksperymencie przeprowadzonym na szczurach dowiedziono, że 2-AG i PEA podczas niedokrwienia mięśnia sercowego zmniejszają w istotny sposób obszar uszkodzenia tkanki [60]. Jednocześnie zmniejszeniu uległo stężenie enzymów uwolnionych z kardiomiocytów – kinazy kreatynowej i dehydrogenazy mleczanowej [60]. Ponadto w mysim modelu niedokrwienia i reperfuzji miokardium wykazano, że stężenie troponiny I pochodzą- cej z kardiomiocytów objętych martwicą jest znacznie mniejsze w przypadku zastosowania agonistów receptorów CB2 [70]. Prawdopodobnie aktywacja receptorów CB2 przyczynia się do „hartowania” serca niedokrwieniem (preconditioning), czyli zabezpiecza kardiomiocyty przed nekrozą w wyniku niedokrwienia oraz następczej reperfuzji [60]. W doświadczeniu przeprowadzonym na myszach, u których wyidukowano zawał mięśnia sercowego dowiedziono, że aktywacja receptorów CB2 zmniejsza migrację leukocytów w miejsce uszkodzonej tkanki [22]. Dochodzi do zmniejszenia możliwości zachodzenia interakcji między leukocytami a śródbłonkiem, a tym samym obniżeniu ulega prawdopodobieństwo wystąpienia dodatkowego uszkodzenia przez zaktywowane komórki układu odpornościowego [22]. Wykazano także, że aktywacja receptorów CB2 przyczynia się do zmniejszonego wytwarzania reaktywnych form tlenu przy zmniejszeniu infiltracji neutrofili w obrębie komór serca po zawale [70].
Endokannabinoidy wykazują właściwości kardioprotekcyjne, ale istnieją dowody na ich negatywny wpływ na układ krążenia. Wykazano, że pobudzenie receptorów CB1 przez AEA w ludzkich komórkach śródbłonka tętnic wieńcowych skutkuje zależną od dawki oraz czasu stymulacją aktywacji szlaków p38 i JNK-MAPK, wzrostem wytwarzania reaktywnych form tlenu oraz śmiercią komórek głównie w wyniku apoptozy [88]. Ponieważ zaktywowane receptory CB1 przez stymulację szlaków p38 i JNK-MAPK promują odpowiedź zapalną, a zaktywowane receptory CB2 wpływając na zmniejszenie ekspresji cytokin prozapalnych hamują chemotaksję i aktywację leukocytów tłumiąc reakcje immunologiczne [88] nasuwa się wniosek: w chwili powstania niedokrwienia mię- śnia sercowego w komórkach śródbłonka zaktywowane receptory CB1 i CB2 pełnią przeciwstawną w stosunku do siebie funkcję – receptory CB1 prozapalną, natomiast receptory CB2 przeciwzapalną. Jednocześnie warto zauważyć, że zastosowanie agonisty receptorów CB2 i/lub antagonisty receptorów CB1 może wpłynąć ochronnie na poddane niedokrwieniu kardiomiocyty. Quercioli i wsp. w badaniu dotyczącym określenia zależności między stę- żeniem endokannabinoidów a występowaniem dysfunkcji w krążeniu wieńcowym u ludzi otyłych [84] stwierdzili, że podwyższone stężenie anandamidu i 2-AG w osoczu wykazuje pozytywną korelację ze zmniejszonym przepływem krwi przez miokardium u osób otyłych w porównaniu do osób o prawidłowej masie [84]. Prawdopodobobnie pod wpływem nadmiernej aktywacji układu endokannabinoidowego dochodzi do nieprawidłowości w funkcjonowaniu śródbłonka i komórek mięśni gładkich tętniczek wieńcowych, co przekłada się na zauważone w doświadczeniu zmiany w przepływie krwi przez miokardium [84].
Podsumowanie
Mimo iż niewiele wiadomo o bezpośrednim wpływie układu endokannabinoidowego na metabolizm mięśnia sercowego, to należy przypuszczać, iż przez ogólny wpływ na metabolizm organizmu przyczynia się do zmian w obrębie miokardium. Z tego też względu upatrywać można nowych możliwości terapeutycznych związanych z udziałem endokannabinoidów w leczeniu licznych zaburzeń serca – nadciśnienia, arytmii, niedokrwienia, zawału, w których dochodzi do nieprawidłowości w zaopatrzeniu energetycznym miokardium. Dlatego też lepsze poznanie bezpo- średniego wpływu tego układu pozwoliłoby kontrolować metabolizm mięśnia sercowego i umożliwiłoby przeciwdziałanie zachodzącym zmianom, które w przypadku mię- śnia sercowego mogą mieć tragiczne skutki.
Otyłość trzewna stanowi główny czynnik ryzyka rozwoju insulinooporności, która może prowadzić do jawnej cukrzycy typu 2 (T2D) z utratą funkcji komórek beta i ostateczną utratą tych komórek. Wydzielanie insuliny przez komórki beta wysp trzustkowych jest ściśle związane z stężeniem glukozy we krwi i regulowane przez wiele mediatorów uwalnianych do krwiobiegu lub miejscowo, w tym endokannabinoidów. Otyłość i jej powikłania, w tym T2D, wiążą się z zwiększoną aktywnością układu endokannabinoidowego/receptora CB1 (CB1R), co potwierdzają efekty terapeutyczne antagonistów CB1R. Podobne korzystne efekty antagonistów CB1R o ograniczonej penetracji do mózgu wskazują na istotną rolę CB1R w tkankach obwodowych, w tym trzustce endokrynnej. Komórki beta trzustki wykazują ekspresję wszystkich składników układu endokannabinoidowego, a endokannabinoidy modulują ich funkcję zarówno poprzez mechanizmy autokrynne, jak i parakrynne. Oddziałują one na wydzielanie insuliny zarówno w warunkach bazalnych, jak i indukowanych glukozą, wpływając również na proliferację i przeżycie komórek beta. Niniejsza krótka recenzja przedstawi dostępne informacje na temat modulacji tych procesów przez endokannabinoidy i receptory, starając się ocenić wkład takich efektów w kontrolę glikemii w T2D i insulinooporności.
Cukrzyca typu 2 (T2D) lub cukrzyca nieinsulinoniezależna to przewlekła choroba charakteryzująca się zaburzoną zdolnością organizmu do metabolizowania glukozy, która często rozwija się u osób otyłych/nadwadze. Otyłość jest czynnikiem ryzyka rozwoju insulinooporności, która definiowana jest jako niemożność normalnej odpowiedzi komórek w tkankach wrażliwych na insulinę na insulinę produkowaną i uwalnianą przez komórki beta wysp trzustkowych w odpowiedzi na wzrost stężenia glukozy we krwi. Powszechnie przyjętym poglądem jest, że u podgrupy otyłych, insulinoopornych jednostek dochodzi do dysfunkcji komórek beta, co prowadzi do zmniejszonej produkcji insuliny, złej regulacji glukozy we krwi, a ostatecznie do T2D [1]. Istnieje także możliwość, że dysfunkcja komórek beta występuje przed lub równolegle do insulinooporności, ponieważ w niektórych przypadkach może być wykrywana znacznie przed wystąpieniem T2D [2].
Komórki beta wysp trzustkowych są wyjątkowo wrażliwe na zmiany stężenia glukozy we krwi. Białko przewoźnika glukozy typu 2 (GLUT-2) umożliwia wejście glukozy do komórek beta, pozwalając tym samym na wyrównanie stężenia glukozy wewnątrzkomórkowej i zewnątrzkomórkowej przez błonę komórkową. Metabolizm glukozy za pomocą glikolizy generuje ATP, a wyższy stosunek ATP/ADP powoduje zamknięcie zależnych od ATP kanałów potasowych (KATP), odpowiedzialnych za utrzymanie potencjału błonowego w spoczynku, uniemożliwiając jonowi potasu przejście przez błonę komórkową. Następujący wzrost dodatniego ładunku wewnątrzkomórkowego prowadzi do depolaryzacji błony komórkowej, co skutkuje otwarciem napięciowo-zależnych kanałów wapniowych i wzrostem stężenia wapnia wewnątrzkomórkowego [3]. Glukozowy sygnał Ca2+i jest zorganizowany w synchroniczny i jednolity wzór oscylacyjny [4], prowadząc do pulsacyjnego wydzielania insuliny [5]. Ostatecznym efektem jest eksport insuliny z komórek beta do krążenia ogólnego.
Układ endokannabinoidowy (ECS) obejmuje receptory kannabinoidowe sprzężone z białkiem G [receptor CB1 (CB1R) i receptor CB2 (CB2R)], ich endogenne ligandy lipidowe lub endokannabinoidy, takie jak arachidonoyletanolamid (AEA) czy anandamid oraz enzymy biosyntezujące i degradowane, jak szczegółowo opisano poniżej. Odkrycie ECS wywołało lawinę eksperymentalnych badań nad jego różnorodnymi funkcjami biologicznymi [6]. Ewidencja zarówno z badań przedklinicznych, jak i badań na ludziach wskazuje, że nadaktywność układu CB1R przyczynia się do rozwoju insulinooporności oraz obu typów cukrzycy, tj. typu 1 (T1D) i typu 2 (T2D) [7,8], a blokada CB1R wykazuje korzystne efekty w łagodzeniu otyłości/zespołu metabolicznego i jego powikłań, w tym T2D [9–11]. Oznacza to, że składniki ECS są obecne i funkcjonalne w tkankach istotnie zaangażowanych w kontrolę glikemii, w tym w trzustce endokrynnej, chociaż nie pojawiła się jeszcze zgoda co do lokalizacji komórkowej i dokładnej funkcji konkretnych składników ECS. W niniejszym artykule przedstawimy krótki przegląd obecnej wiedzy na temat roli ECS w funkcji i przeżyciu komórek beta, w związku z jego zaangażowaniem w T2D.
Najnowsze badania w dziedzinie biomedycyny wykazały integralną rolę układu endokannabinoidowego (ECS) w określaniu ryzyka sercowo-metabolicznego organizmu ludzkiego. Mechanizm ten jest pośredniczony poprzez wiązanie endokannabinoidów z receptorami CB1. Uważa się, że stymulacja receptorów CB1 w mózgu kontroluje i wpływa na apetyt. W normalnej fizjologii aktywacja receptorów CB1 odpowiada za homeostazę energetyczną, reguluje emocje oraz zachowania, takie jak lęk, strach, apetyt, spożycie jedzenia i wody. Receptory CB1 znajdują się również w tkankach obwodowych, takich jak wątroba, trzustka, mięśnie szkieletowe i tkanka tłuszczowa, odgrywające istotną rolę w metabolizmie lipidów i glukozy. Nadmierna aktywacja ECS wiąże się z różnymi chorobami metabolicznymi, takimi jak dyslipidemia, insulinooporność, lipogeneza, nadmierne przybieranie na wadze i zwiększenie otyłości brzusznej. Wszystkie te zdarzenia prowadzą do zwiększonego ryzyka sercowo-naczyniowego. Stosowanie selektywnego blokera receptora CB1, takiego jak rimonabant, wykazało redukcję obwodu talii, lepszą kontrolę glikemii, obniżenie poziomu triglicerydów, podniesienie poziomu cholesterolu HDL i ogólną redukcję całkowitej masy ciała. Lek ten został zalecany dla pacjentów z zespołem metabolicznym.
Układ endokannabinoidowy (ECS) reguluje homeostazę komórkową i metabolizm całego organizmu. Istnieje autonomiczny ECS w trzustce endokrynnej, obejmujący receptor kannabinoidowy 1 (CB1R), który występuje w komórkach β. W tym kontekście omawiamy konflikty związane z funkcją(-ami) endokannabinoidów w trzustce endokrynnej, które wywołały zamieszanie przy definiowaniu roli ECS w wyspach trzustkowych, zwłaszcza roli(-i) CB1R w komórkach β. Omawiamy również najnowsze opublikowane dane dotyczące ECS w wyspach trzustkowych. CB1R w szczególności nie jest wyłącznie negatywnym modulatorem wydzielania insuliny, ponieważ uczestniczy także w intra-wyspowym zapaleniu podczas spożycia dużej ilości tłuszczu i cukru oraz działa jako negatywny regulator przeżywalności i wymiany komórkowej β. Rozważamy również możliwość wykorzystania CB1R jako celu terapeutycznego w leczeniu cukrzycy.
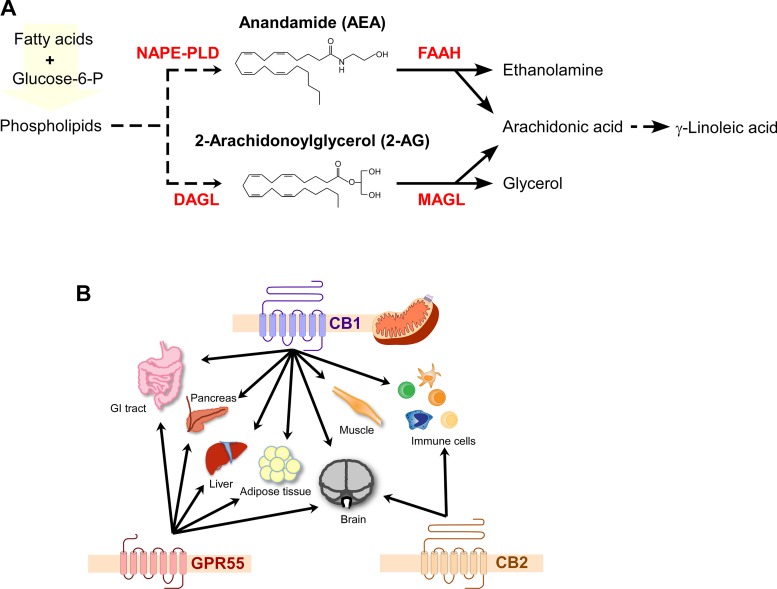
Układ endokannabinoidowy (ECS) to starożytna sieć sygnałowa zaangażowana w utrzymanie homeostazy komórkowej (15). Składa się z dwóch endokannabinoidów (EC), anandamidu (AEA) i 2-arachidonoyloglicerolu (2-AG); ich receptorów sprzężonych z białkiem G, kannabinoidowych 1 (CB1R) i 2 (CB2R); niestandardowego GPR55; oraz enzymów odpowiedzialnych za syntezę i degradację EC (34, 35) (Ryc. 1). EC są syntetyzowane na żądanie przez enzymy N-acylofosfatyloetanolaminy-fosfolipaza D (NAPE-PLD; syntaza AEA) i diacyloglicerol lipaza (DAGL; syntaza 2-AG) z związków prekursorowych na bazie arachidonianu związanych z błoną komórkową (34). Po wydzieleniu i internalizacji, EC są szybko degradowane przez hydrolazę amidową kwasów tłuszczowych (FAAH) i monoacyloglicerolową lipazę (MAGL), a produkty rozkładu są ponownie przetwarzane (34) (Ryc. 1A). W otyłości, jak to ma miejsce w przypadku spożywania diety zachodniej bogatej w tłuszcze i cukry, ECS staje się nadaktywny, głównie poprzez zwiększenie syntezy EC (8, 39).
Rysunek 1. Układ endokannabinoidowy (ECS). A: schemat syntezy i degradacji endokannabinoidów (EC). B: receptory kannabinoidowe (CBR) obejmują klasyczne receptory kannabinoidowe 1 i 2 (CB1R i CB2R) oraz niestandardowy receptor sprzężony z białkiem G 55 (GPR55), które są obecne na błonie komórkowej. Dodatkowo, CB1R jest również obecny w zewnętrznej błonie mitochondrialnej w niektórych tkankach. Wszystkie trzy receptory CBR są ekspresowane w mózgu. Na obwodzie CB1R i GPR55 są ekspresowane w tkankach, jak pokazano.
Receptory kannabinoidowe (CBR) są obecne na błonie komórkowej wielu narządów (rysunek 1B), regulując szereg funkcji. Podczas gdy CB2R występuje głównie w komórkach układu immunologicznego (4, 18), CB1R jest obfity w mózgu, gdzie kontroluje presynaptyczną hamulację zwrotną pobudzenia oraz reguluje apetyt i reakcję nagrody w podwzgórzu. Na obwodzie reguluje motorykę jelit (52) i wydzielanie incretin [glukozozależnego peptydu insulinotropowego (GIP) i peptydu podobnego do glukagonu (GLP-1)] w jelitach. CB1R jest wyrażany w innych tkankach o funkcji endokrynnej, takich jak nadnercza, jajniki, jądra i nasieniowody (10). W trzustce endokrynnej aktywacja CB1R przez autonomiczną syntezę EC i autokrynne działanie w β-komórkach pełni funkcję sprzężenia zwrotnego negatywnego dla wielu funkcji β-komórkowych (19, 26).
CBR są sprzężone z białkiem Gαi/o i hamują aktywację adenylanu cyklazy (AC) oraz aktywność cAMP-PKA. Aktywują również MAPK i hamują kanały wapniowe typu L, N i P/Q oraz kanały potasowe o prostokątnej charakterystyce wewnętrznej, prowadząc do hamowania przekazu sygnału i zmniejszonego uwalniania produktów wydzielniczych z β-komórek (19, 20). W wątrobie i β-komórkach aktywacja CB1R ujemnie wpływa na szlak receptora insuliny (IR) poprzez bezpośrednią interakcję jej podjednostki Gαi z podjednostką β IR (27), a osłabiona aktywacja CB1R poprawia działanie insuliny (12, 33). Przeanalizujemy literaturę w kontekście modulacji funkcji wysp trzustkowych.
Konflikty — Brzydkie.
Konflikt 1. Które komórki wysp trzustkowych zawierają receptory kannabinoidowe (CBR), a które syntetyzują endokannabinoidy (EC)? Brak specyficzności przeciwciał doprowadził do sprzecznych doniesień dotyczących obecności CBR w różnych komórkach wysp trzustkowych. Dowody wskazują, że ECS nie jest wyrażony ani w tkance zrazikowej, ani przewodowej dojrzałej trzustki. W trzustce endokrynnej CB1R był różnie zgłaszany w komórkach α-, δ- i β-komórkach (7, 24, 26, 43, 56). Ostatnio wykazaliśmy, analizując molekularnie pojedyncze komórki rozproszone z ludzkich wysp, że CB1R jest wyrażany we wszystkich β-komórkach, ale nie w komórkach α- lub δ- (19). My i inni również zgłosiliśmy, że izolowane wyspy zawierają autonomiczny ECS (7, 26): EC są syntetyzowane na żądanie w odpowiedzi na glukozę w sposób zależny od stężenia, przy czym 2-AG jest najbardziej obfitującym EC (7, 26, 39). W trakcie rozwoju EC wpływają na ostateczną architekturę dorosłych wysp (38), co wskazuje na rolę rozwojową ECS w wyspach.
Konflikt 2. Czy istnieją różnice między ekspresją CBR w wyspach u gryzoni a ludzi, które dodają niepewności? Ludzki gen CB1R koduje trzy różne izoformy białek o zróżnicowanej powinowactwie do ligandu i tkankowym rozmieszczeniu (19, 49, 53, 55). Ludzkie β-komórki i hepatocyty obficie wyrażają CB1b, krótszą izoformę praktycznie nieobecną w mózgu (19). Istnienie izoform, które mogą być preferencyjnie ukierunkowane, wprowadza nową zmienną do równań terapeutycznych. Podejścia farmakologiczne do ukierunkowywania aktywności CB1R w obwodzie były problematyczne ze względu na lipofilową naturę modyfikatorów CBR, które swobodnie przenikają barierę krew-mózg.
Konflikt 3. Jaki jest dowód na obecność innych CBR oprócz CB1R w wyspach? Całe wyspy zawierają makrofagi, naczynia krwionośne i zakończenia nerwowe oraz wykazują transkrypty dla CB2R, ale w znacznie mniejszym stopniu (różnica 100-krotna) niż CB1R (7, 16, 24). Syntetyczny antagonist CB2R AM630, w ilościach farmakologicznych, zmniejszał EC-indukowaną redukcję oscylacji stężenia Ca2+ wewnątrz wysp w sposób zależny od toksyny pertusisowej (24). Syntetyczny agonista CB2R JWH133 zmniejszał wydzielanie insuliny z izolowanych mysich i ludzkich wysp (7, 24), podczas gdy inny syntetyczny agonista CB2R JWH015, zwiększał wydzielanie insuliny stymulowane glukozą z izolowanych mysich wysp (57). U myszy JWH133 zmniejszał poziomy glukozy we krwi po obciążeniu glukozą, podczas gdy AM630 wywoływał przeciwny efekt (6). W związku z tym istnieje konflikt, zwłaszcza między wynikami in vivo a in vitro przy użyciu środków modyfikujących CB2R. GPR55 jest silnie wyrażany w mózgu (50) i sygnalizuje poprzez białka Gα13 i Gq (29). Odkryto wiele związków endogennych i syntetycznych, w tym kannabinoidy, takie jak THC i AEA w stężeniach nanomolowych (2, 48), które związują się z nim. Jego endogenne ligandy wydają się być lizofosfatydyloinozytolem i jego pochodną 2-arachidonoyloinozytolem. Na obwodzie wykazano, że jest wyrażany w białej tkance tłuszczowej, wątrobie, przewodzie pokarmowym i wyspach (21, 46) (rysunek 1B). Aktywacja GPR55 przez O-1606, syntetyczny agonista, zwiększała mobilizację stężenia Ca2+ stymulowaną glukozą w β-komórkach i wydzielanie insuliny, a na podstawie immunohistochemii, GPR55 kolokalizuje się z barwieniem insulinowym (46). Inne agonisty GPR55, takie jak Abn-CBD, miały zdolność zwiększenia wydzielania insuliny z linii komórkowej insulinoma (40). In vivo, u gryzoni, aktywacja GPR55 prowadzi do poprawy tolerancji glukozy i zwiększenia poziomów glukozy stymulowanej glukozą insuliny (40, 46). GPR55 bierze udział nie tylko w funkcji β-komórkowej, ale, podobnie jak CB1R, również utrzymaniu masy β-komórkowej. Blokada GPR55 zmniejszała proliferację i przeżycie komórek oraz powodowała przesunięcie metaboliczne ku fosforylacji oksydacyjnej, zmniejszając produkcję mleczanu i karnityny oraz sygnalizację PI3K-Akt (9). U myszy z cukrzycą wywołaną streptozotocyną, leczenie Abn-CBD zwiększało liczbę β-komórek w wyspach, obniżało poziomy glukozy we krwi i zwiększało poziomy insuliny; te efekty były zależne od obecności GLP-1R i GIPR, ponieważ myszy z ogólnoustrojowym wyłączeniem tych receptorów nie wykazywały zmian w poziomach glukozy ani insuliny po leczeniu Abn-CBD w porównaniu z pojazdem (41). Jakikolwiek wpływ fizjologicznych poziomów endogennych ligandów na aktywność GPR55 w β-komórkach nie został przedstawiony. Należy zauważyć, że opisano heterodimery rodziny receptorów kannabinoidowych, które wpływają na ich sygnalizację wewnątrzkomórkową (3, 11, 25). Istnienie tych interakcji, chociaż jeszcze nie opisanych w β-komórkach, może komplikować badania poszczególnych receptorów: przyszłe badania takich możliwych interakcji są uzasadnione.
Konflikt 4. Czy endokannabinoidy są promiskuityczne, czy istnieje jeden ligand dla jednego receptora? AEA ma wyższe powinowactwo do receptora CB1 niż do receptora CB2, podczas gdy 2-AG łączy się z obiema receptorami w jednakowym stopniu. Jednak endokannabinoidy zdają się także aktywować inne receptory, w tym GPR55, w zakresie nanomolowym (45). Przyjmując, że wszystkie trzy receptory są obecne w komórkach wysp trzustkowych, stosowanie egzogennych endokannabinoidów do badania specyficznej roli CB1R może nie być najlepszym podejściem. Istnieją bardziej konkretne alternatywy przy użyciu syntetycznych ligandów, takich jak ACEA, choć istnieją pewne, takie jak AM251, SR141716A lub nowa substancja LH-21, które są znanymi antagonisami CB1R, a jednocześnie wykazują aktywność agonistyczną wobec GPR55 (14, 42, 45, 47). Ponadto istnieją różnice w powinowactwie między gatunkami (55), które należy wziąć pod uwagę przy badaniu ich wpływu na wyspy trzustkowe u ludzi w porównaniu z gryzoniami. Ostatnie dane pokazują, że inne receptory, takie jak receptor potencjału TRPV1, aktywowane przez kannabinoidy (58), są zaangażowane w regulację poziomu Ca2+ w komórkach, co wpływa na funkcję wysp trzustkowych (1, 36). Co ważne, komórki β- i α-trzustki wykazują ekspresję kluczowych enzymów do metabolizmu AEA (37, 54), a stosowanie kultur komórkowych mieszanych populacji lub stosowanie całości wysp nie może dostarczyć pełnego obrazu roli receptora CB1 w komórkach β, ani, rzeczywiście, roli dowolnego indywidualnego receptora. Ponadto znaczne ilości krążącej AEA znajdują się we krwi (39), co może wprowadzić zamieszanie w wynikach dotyczących endogennych endokannabinoidów w wyspach trzustkowych. Endokannabinoidy nie tylko sygnalizują poprzez różne receptory, ale również poprzez różne podjednostki receptorów kanabinoidowych. Oprócz sygnalizowania poprzez podjednostkę Gαi, kilka ligandów, w zakresie mikromolowym, sygnalizuje poprzez podjednostkę Gq receptora CB1 w neuronach hipokampu, która łączy się z fosfolipazą C w celu uwolnienia Ca2+ z wnętrza komórki i powodzenia depolaryzacji (28). W rzeczywistości w komórkach wysp trzustkowych mikromolarne stężenia syntetycznych i endogennych agonistów receptora CB1 indukują wydzielanie insuliny (30–32), co wskazuje, że suprafizjologiczne stężenia kannabinoidów mogą także sygnalizować poprzez podjednostkę Gq w komórkach β.
Dysfunkcja komórek β — złe
Wczesne badania skłoniły nas do wniosku, że aktywowane receptory CB1 są jedynie negatywnymi regulatorami AC, zmniejszającymi bodźce do wydzielania insuliny, takie jak inkretyny i pobudzający adenyloniany cyklazę peptyd przysadkowy, które zależą od aktywacji AC (Fig. 2). Jednakże pojawia się bardziej złożony obraz, implicujący wpływ na kanały jonowe K+ i Ca2+, sygnalizację MAPK, syntezę ceramidów, funkcję mitochondriów oraz sygnalizację Akta (20, 27). Ponadto CB1R w komórkach β wpływa na żywotność komórkową. Poprzez bezpośrednią interakcję aktywowanego receptora CB1 z receptorami insuliny, fosforylacja Akta i Bad są zmniejszone (27), prowadząc do zmniejszonej proliferacji i zwiększonej śmierci komórkowej β. W przeciwieństwie do tego, usunięcie CB1R w komórkach β zapobiegało indukowanej dietą produkcji oksydacyjnego stresu wewnątrz wysp trzustkowych i redukowało aktywację szlaku MAPK, co zmniejszało aktywację Nlrp3 inflamasomu i rozpad kaspazy 3 indukowane wysokim stężeniem glukozy i palmitynianu, co prowadziło do zachowania żywotności wyspy (20) (Fig. 2). Ponadto CB1R jest zaangażowany w pośredni sposób w żywotność wysp: CB1R na miejscowych makrofagach wysp i trzustki, gdy jest aktywowany, aktywuje inflamasom Nlrp3 i zwiększa wydzielanie IL-1β z makrofagów; w ten sposób uczestniczy w indukowanej dietą zapalnej odpowiedzi wyspy i dysfunkcji komórkowej β (22, 23). W zachodnich dietach występuje stała stymulacja komórek β z powodu utrzymującej się dysglykemii. Nieustanne bodźce prowadzą nie tylko do zwiększenia wydzielania insuliny, ale także do wzrostu lokalnych poziomów endokannabinoidów. Ponadto podwyższone poziomy endokannabinoidów, poprzez stymulowanie receptora CB1 w makrofagach i komórkach β, aktywują odpowiedź zapalną, powodując dysfunkcję komórkową β i apoptozę.
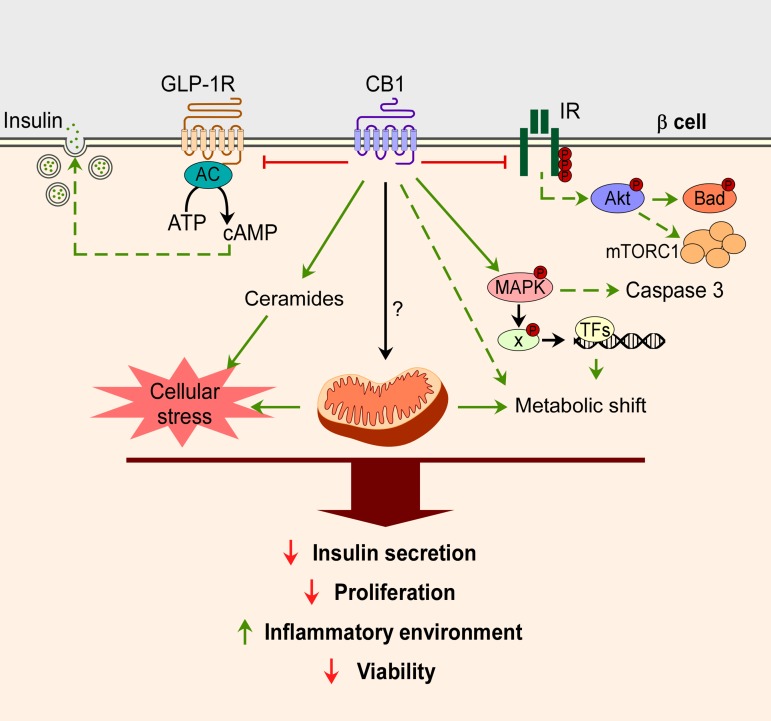
Rysunek 2.
Działania receptora kanabinoidowego 1 (CB1R) w komórkach β. Aktywacja CB1R powoduje obniżenie aktywacji receptora peptydu przysadkowego przypominającego glukagon (GLP)-1 i receptora insuliny (IR), co z kolei zmniejsza GLP-1-śródwydzielane wydzielanie insuliny, a także redukuje fosforylację Aktu i jego szlaki sygnalizacyjne. Aktywacja CB1R aktywuje szlak MAPK i indukuje rozpad kaspazy 3 oraz ekspresję czynników transkrypcyjnych, które prowadzą do przesunięcia metabolizmu w komórkach β. Bezpośrednie lub pośrednie działanie CB1R na mitochondria w komórkach β pozostaje do ustalenia, ale wraz z indukowaniem syntezy ceramidów przez CB1R, przesunięcie metabolizmu prowadzi do zwiększonego wewnątrzkomórkowego stresu oksydacyjnego. Ogółem, chroniczne nadaktywowanie układu endokannabinoidowego (ECS) w komórkach β prowadzi do ograniczonej funkcji, tj. zmniejszonego wydzielania insuliny, zmniejszonej proliferacji, aktywowanych makrofagów miejscowych i zmniejszonej żywotności komórek β.
Dobre
Omówiliśmy, że komórki β zawierają autonomiczny układ endokannabinoidowy (ECS), a endokannabinoidy (ECs) są wytwarzane na żądanie w odpowiedzi na stymulację glukozy i działają autokrynnie. Ten rodzaj sygnalizacji działa jako ujemna informacja zwrotna, aby uniknąć hipoglikemii i utrzymać homeostazę w wydzielaniu insuliny. Ponadto wydaje się, że ECS w komórkach β ewoluował, aby chronić przed stanem zapalnym, który potencjalnie byłby szkodliwy dla komórek β. CB1R w komórkach β pełni funkcję broni dwuobrotowej: 1) gdy jest aktywowany ostrożnie, chroni przed nadmierną aktywnością komórek β, 2) ale w stanie otyłości, ECs w krążeniu i w wyspach trzustkowych są chronicznie zwiększone, a ich aktywacja CB1R — nawet gdy komórki β nie są w stanie poposiłkowym — w końcu utrudnia funkcję komórek β, prowadząc do zapalenia wysp trzustkowych i zwiększonej apoptozy komórek β. Antagonizm CB1R redukuje zapalenie wysp trzustkowych indukowane dietą (22, 23, 47), zwiększa fosforylację Aktu i Bad, oraz zwiększa sygnalizację mTORC1 (5, 27), tym samym sprzyjając odnowie komórek β i ich przeżywalności oraz zapobiegając apoptozie, co może być wykorzystane jako terapia w leczeniu cukrzycy. Jednak aktywacja receptorów kannabinoidowych (CBRs) jest skuteczną terapią w niektórych rodzajach nowotworów, w zależności od ekspresji receptora, ponieważ aktywacja CB1R może zmniejszać proliferację komórkową i indukować apoptozę (44). Indywidualne i tkankowe terapie będą musiały zostać wzięte pod uwagę przy rozważaniu ECS jako celu terapeutycznego.
Wnioski.
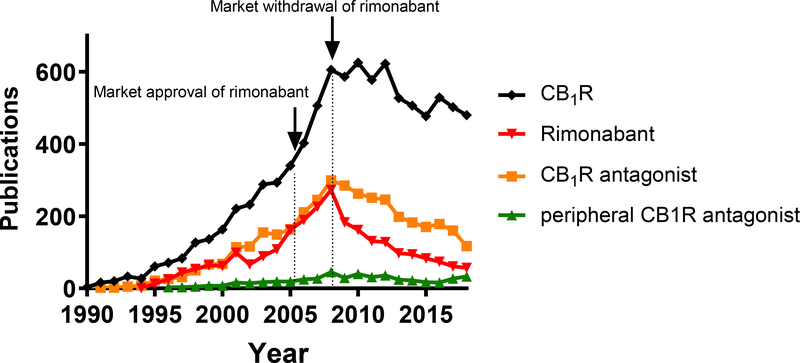
ECS to potencjalny cel dla nowych terapii chroniących wyspy trzustkowe przed zapaleniem i apoptozą. Wczesne lata 2000 roku przyniosły dostępność rimonabantu do leczenia otyłości (13, 17, 51), i mimo osiągnięcia obiecujących wyników metabolicznych, pojawiły się niekorzystne efekty psychiatryczne. Być może efekty te można pokonać dzięki drugiej generacji syntetycznych modyfikatorów CB1R, które nie przenikają przez barierę krew-mózg. Ponadto izoformy CB1R mogą być nowymi celami dla projektowania leków, w tym oligonukleotydy morfolino do celowania w konkretne warianty skrawków. Rozważać można również zmiany w rodzajach tłuszczów w diecie, aby zmniejszyć syntezę EC z prekursorów.
Endokannabinoidy działające poprzez receptory CB1 (CB1R) odgrywają kluczową rolę w regulacji homeostazy energetycznej, co stanowiło podstawę do opracowania farmaceutycznego antagonistów CB1R w leczeniu otyłości. Pomimo skuteczności pierwszego w swojej klasie antagonisty CB1R, rimonabantu, w łagodzeniu otyłości i jej wielokrotnych powikłań kardiometabolicznych, został on wycofany z użycia klinicznego ze względu na działania niepożądane neurofizjatryczne ośrodkowego układu nerwowego, co zatrzymało dalszy rozwój terapeutyczny tej klasy związków. W porównaniu do mózgu receptory CB1R są wyrażane na niskim, ale funkcjonalnym poziomie w narządach obwodowych zaangażowanych w regulację homeostazy energetycznej, takich jak wątroba, mięśnie szkieletowe, tkanka tłuszczowa i trzustka. W ostatnich badaniach przedklinicznych selektywne ukierunkowanie tych receptorów przez „antagonisty CB1R drugiej generacji” o ograniczonym oddziaływaniu na ośrodkowy układ nerwowy replikowało korzyści metaboliczne rimonabantu w modelach myszy otyłych i chorych na cukrzycę, bez wywoływania efektów ubocznych związanych z ośrodkowym układem nerwowym. Zwiększona aktywność CB1R przyczynia się także do złożonych, wieloczynnikowych zaburzeń, takich jak różne formy zwłóknienia tkanek, leczenie których może korzystać z jednoczesnego zaangażowania więcej niż jednego celu terapeutycznego. W związku z tym nowe „antagonisty CB1R trzeciej generacji” stanowiące hybrydowe inhibitory CB1R i indukowalnej syntazy tlenku azotu zostały przetestowane w modelach myszy z marskością wątroby i płuc, gdzie ich skuteczność przeciwfibrotyczna okazała się większa niż skuteczność leków hamujących tylko jeden z tych celów. W tej recenzji omówimy wyzwania i możliwości, jakie stwarzają antagonisty CB1R drugiej i trzeciej generacji, oraz ich potencjalne zastosowania terapeutyczne.
Odkrycie receptorów kannabinoidów sprzężonych z białkami G i ich endogennych ligandów we wczesnych latach 90. XX wieku doprowadziło do gwałtownego wzrostu informacji na temat ich roli w różnych fizjopatologiach i, co za tym idzie, jako potencjalnych celów farmakoterapeutycznych. Rola endokannabinoidów działających poprzez hipotalamiczne receptory kannabinoidowe typu 1 (CB1R) jako czynników pobudzających apetyt, oreksyjnych (Di Marzo et al., 2001), była podstawą do opracowania farmaceutycznego antagonistów CB1R w leczeniu otyłości i jej powikłań metabolicznych. W kilku wieloośrodkowych badaniach klinicznych III fazy, pierwszy w swojej klasie antagonist/inwersyjny agonista CB1R, rimonabant, nie tylko spowodował utratę wagi u osób z zespołem metabolicznym, ale także istotnie poprawił liczne parametry kardiometaboliczne zmienione w otyłości, takie jak obwód talii, hemoglobina A1c, HDL, cholesterol i triglicerydy w osoczu (Despres et al., 2005; Van Gaal et al., 2008). W tym samym czasie prowadzono wiele badań klinicznych nad antagonistami CB1R (taranabant, otenabant, ibipinabant) (Tabela 1). Pomimo zatwierdzenia rimonabantu do użytku klinicznego w około 40 krajach, niepożądane efekty neuropsychiatryczne ośrodkowego układu nerwowego, takie jak lęk i zwiększona skłonność samobójcza (Christensen et al., 2007), spowodowały jego wycofanie z rynku w 2008 roku. To zatrzymało dalszy rozwój farmaceutyczny całej tej klasy związków (Tabela 1) oraz przerwało rozwój badań związanych z receptorami CB1R (Rysunek 1).
Rysunek 1. Skutki wycofania rimonabantu z rynku dla badań związanych z receptorem CB1R.
Narastające dowody wskazują, że nadmiernie aktywny układ endokannabinoidowy (ECS) może przyczyniać się do rozwoju cukrzycy, sprzyjając pobieraniu i gromadzeniu energii, zaburzając zarówno metabolizm glukozy, jak i lipidów, wywierając proapoptotyczne działanie w komórkach beta trzustki oraz ułatwiając stan zapalny w wyspach trzustkowych. Ponadto hiperkalcemia związana z cukrzycą również została wskazana jako czynnik wywołujący zakłócenia w ECS, nasilając procesy patologiczne wspomniane wcześniej, co ostatecznie prowadzi do błędnego kręgu. Przekonujące dowody z badań przedklinicznych wskazują, że ECS wpływa również na cukrzycowo-indukowany stres oksydacyjny, stan zapalny, włóknienie i następujące po nich uszkodzenia tkanek w narządach docelowych powikłań cukrzycowych. W tym przeglądzie przedstawiamy aktualizację dotyczącą roli ECS w patogenezie cukrzycy oraz mikronaczyniowych powikłań cukrzycowych (retinopatii, nefropatii i neuropatii) i powikłań sercowo-naczyniowych. Omawiamy również potencjał terapeutyczny związany z ukierunkowywaniem ECS.
Główny psychoaktywny składnik Cannabis sativa, Δ9‐tetrahydrocannabinol (THC), został zidentyfikowany 50 lat temu. Od tego czasu wiele wysiłku zostało skierowane na identyfikację endogennych związków, których działanie biologiczne jest naśladowane przez THC, oraz na wyjaśnienie ich roli w różnych procesach fizjologicznych i patologicznych. Układ endokannabinoidowy (ECS) obejmuje endokannabinoidy (ECs), enzymy regulujące ich produkcję i degradację oraz receptory, przez które przekazują sygnały. Anandamid (AEA) i 2-arachidonoylglicerol (2-AG), najbardziej zbadane endokannabinoidy, to biologicznie aktywne mediatory lipidowe produkowane z fosfolipidów błony komórkowej. ECs są syntezyowane „na żądanie”, AEA głównie poprzez hydrolizę N-arachidonoylofosfatydyloetanolaminy przez fosfolipazę D, a 2-AG z diacyloglicerolu przez diacyloglicerol lipazę (DGL), choć istnieją również równoległe ścieżki biosyntezy. Po wytworzeniu, AEA lub 2-AG są natychmiast uwalniane, aby oddziaływać na swoje receptory, a następnie szybko degradowane przez hydrolazę amidu kwasu tłuszczowego lub lipazę monoacyloglicerolową (MGL) odpowiednio. Efekty ECs są przekazywane głównie przez sprzężone z białkiem Gi/o receptory kannabinoidowe 1 lub 2 (receptor CB1/receptor CB2), z możliwym zaangażowaniem dodatkowych receptorów, takich jak GPR-55. AEA sygnalizuje głównie za pośrednictwem receptorów CB1, podczas gdy 2-AG jest pełnym agonistą zarówno receptorów CB1, jak i CB2. Aktywacja receptora prowadzi do różnorodnych odpowiedzi biochemicznych, w tym hamowania kanałów wapniowych zależnych od napięcia i aktywności adenylanu cyklazy, prowadząc do obniżenia poziomu cAMP, a także aktywacji kanałów K+, fosfolipaz i szlaków MAPK, te ostatnie za pośrednictwem mechanizmów niezależnych od białka G (Howlett et al., 2010; Horváth et al., 2012).
Używamy plików cookies w celu optymalizacji naszej witryny i naszych serwisów. Więcej informacji:
Funkcjonalne
Zawsze aktywne
Przechowywanie lub dostęp do danych technicznych jest ściśle konieczny do uzasadnionego celu umożliwienia korzystania z konkretnej usługi wyraźnie żądanej przez subskrybenta lub użytkownika, lub wyłącznie w celu przeprowadzenia transmisji komunikatu przez sieć łączności elektronicznej.
Preferencje
Przechowywanie lub dostęp techniczny jest niezbędny do uzasadnionego celu przechowywania preferencji, o które nie prosi subskrybent lub użytkownik.
Statystyka
Przechowywanie techniczne lub dostęp, który jest używany wyłącznie do celów statystycznych.Przechowywanie techniczne lub dostęp, który jest używany wyłącznie do anonimowych celów statystycznych. Bez wezwania do sądu, dobrowolnego podporządkowania się dostawcy usług internetowych lub dodatkowych zapisów od strony trzeciej, informacje przechowywane lub pobierane wyłącznie w tym celu zwykle nie mogą być wykorzystywane do identyfikacji użytkownika.
Marketing
Przechowywanie lub dostęp techniczny jest wymagany do tworzenia profili użytkowników w celu wysyłania reklam lub śledzenia użytkownika na stronie internetowej lub na kilku stronach internetowych w podobnych celach marketingowych.